C2005/F2401 '10 Lecture #22
© Copyright 2010
Deborah Mowshowitz and Lawrence Chasin Department of Biological Sciences
Columbia University New York, NY
Last Updated:
12/06/2010 03:56 PM
Handouts: 22 A (Dihybrid Crosses) and 22 B (Independent Assortment & Linkage).
I. Pedigrees. See handout 21 B & notes of last lecture.
II. Crosses with Multiple genes (on separate chromosomes) -- Genotypes
A. Consider a dihybrid cross (for example) AABB X aabb. What will the offspring (F1) be?
1. Terminology: A monhybrid cross (AA X aa) gives an F1 that is hybrid for 1 gene (Aa). A dihybrid cross (AABB X aabb or AAbb X aaBB) gives an F1 that is hybrid for 2 genes (AaBb), and so on.
2. Procedure: Same procedure as with monohybrid crosses (such as AA X aa) -- figure out the gametes, and mix them to get zygotes.
3. Results: In this case (AABB X aabb), first parent must produce AB gametes and second parent ab gametes, so all zygotes must be AaBb = F1. (If you think gametes of first parent could be AA or BB, you should convince yourself why you are wrong. Put the genes on homologous chromosomes, go through meiosis, and see why only AB gametes are possible.)
B. What will gametes of AaBb be? See handout 22B
Suppose you want to cross the F1's (AaBb) from above to get the F2. How do you do it? Repeat standard procedure as in previous cases. Figure out gametes, then zygotes. So let's figure out the gametes of AaBb. What do you expect? See handout 22B or Becker fig. 20-15 [20-16].
Let's start with the simplest case: Independent Assortment/Separate Chromosomes. (See right panel of Handout 22B, or Becker fig.20-14 (20-15) or Sadava fig. 12.8 (10.8). Other cases are considered later.
C. What will the zygotes be for the case of independent assortment? See Handout 22A.
1. Using a Square: Can make a 4 X 4 Punnett square with gametes (AB Ab, aB, ab) on each side and fill it all in. (See Sadava fig. 12.7 (10.7) for an example.) This method is tedious, subject to error, and unwieldy when crosses involve more than 2 genes. In order to use this method to get the gametes and do the square, we have to assume that the two genes assort (are distributed) independently. If this is so, there is an easier way to figure out the offspring -- the branch method.
2. Using the Branch/probability method: This is another way to figure out what zygotes are expected, and in what proportions. This method works for any number of genes as long as they assort independently. This is the "branch" method (shown on your handout; it is just the application of elementary probability).
a. Figure out what would happen if you did the cross with one gene (here Aa X Aa). Use a small square if you don't remember. In this case results will be three genotypes, AA, Aa, aa in ratios of 1:2:1.
b. Figure out what would happen if you did the cross with the other gene(s), here Bb X Bb.
c. Put the results together.
(1) Number of possible different genotypes: Since the alleles of the
alpha gene and the alleles
of the beta gene are distributed independently, any expected alpha genotype can go with any
expected beta genotype. This gives the following possibilities:
AA with BB, Bb or bb; Aa with BB, Bb or bb; aa with BB, Bb, or bb. (This is most easily
followed using a branching diagram as shown on the handout.) This shows that 9 different
genotypes are expected; the square gives the same result.
(2)
Proportions of different genotypes:
Figure out the chance of each alpha combination (AA, Aa, or aa) and of each beta combination; chance of any particular combination (say aaBb)
is the product of the individual chances (= 1/4 X 1/2 for aaBb).
The general rule is: if the chances of any two (alternative) outcomes are known, the
chance of both happening at the same time is their product .
d. Important: Don't panic if probability is not your strong point. All the probability you need to handle genetics (at the level of this course) is as follows:
*Note: the "or means add" rule only works if the two alternatives are mutually exclusive. For example, you can't be both AA and Aa at the same time, so chance of being one or the other is the sum of the two chances. If the alternatives can both occur, the rule is different. (Famous example: Suppose there is a chance of 50% that it will snow today, and a 50% chance tomorrow. What is the chance it will snow either today or tomorrow? The answer is not 100%!)
e. How this works for crosses with many genes. Suppose you have a cross involving more than two genes, such as AaBBccDd X aaBbCCDd. If all genes assort independently, you can figure out the results for each gene (Aa X aa; BB X Bb etc.) and multiply the probabilities to figure out the chances of getting some combination such as AaBbCcDd. In this case, that would be 1/2 X 1/2 X 1 X 1/2.
To review independent assortment, try problem 10-1 (A-C).
III. Crosses with Multiple genes -- Phenotypes
A. One gene -- One Trait. Two genes & two traits overall, but each gene controls only one trait or characteristic. (For example, say gene alpha controls eye color and gene beta controls the ability to digest galactose.)
Assume the simplest case -- complete dominance of A over a and B over b; each gene affects a different trait. (A sample set up, as shown on handout 22A, would be A → one enzyme; B → different enzyme; each enzyme acts in a different pathway.)
1. How many phenos? In the simplest case, there are 4 different phenotypes corresponding to A_B_; A_bb; aaB_ and aabb. A_B_ gets both the alpha job and the beta job done, aabb does neither and so on.
2. How many of each pheno?
a. For gene alpha: In this case, you need to know what proportion are A_ (= what is chance of getting A_)?
A_ = AA plus Aa.
If chances of AA and Aa are known, chance of either one (of A_) = sum of the two chances.
From Aa X Aa, 1/4 should be AA and 1/2 Aa
How many A_ ? Chance of A_ = 1/4 + 1/2 = 3/4
How many aa? 1/4 expected from Aa X Aa. Alternatively, we know 3/4 are A_ and everyone else is aa.b. For gene beta: By the same reasoning, 3/4 should be B_ and 1/4 bb.
c. For both genes:
What proportion should be A_B_? That's 3/4 X 3/4 = 9/16.
What proportion A_bb? 3/4 X 1/4 = 3/16.
Using this reasoning, the proportions of the 4 phenotypes should be 9/16: 3/16:3/16:1/16.
This gives the famous ratio of 9:3:3:1.
To say it another way,
9/16 should be able to do both jobs
3/16 able to do the beta job or alpha job (but not the other)
1/16 should be able to do neither.
To review the genotypes and phenotypes expected in cases like this (independent assortment, and one gene/one trait) try 10-1 part D, 10-2, 10-3 & 10-5.
B. One gene -- Two (or many) traits.
Suppose one gene → 1 peptide. This one peptide can have multiple direct or indirect effects. This is called pleiotropy. In this case, one gene affects several traits. (For an example, see Waardenburg syndrome, described in problem 10-18.)
C. Two (or more) genes -- One Trait.
Suppose 2 genes → 2 peptides. Both genes/peptides can affect the same trait, for example by affecting the same or different steps in a single pathway. Examples:
1. Two genes control a single step.
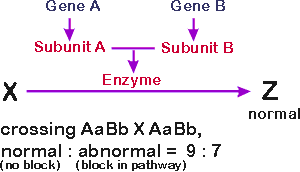
Suppose 2 different peptides → 1 enzyme (with two subunits). Enzyme catalyzes, say, X → Z. (See picture below.)
Then an absence of either gene product will cause a block in the same step (X to Z). Therefore, aa B_, A_bb and aabb will have the same phenotype - - in all these cases, X to Z will be blocked, so X will not be broken down and/or Z will not be made.
If you do the cross AaBb X AaBb, the ratio of normal to mutant phenotypes will be 9:7.
If X is colorless and Z is colored, say black, then phenotypic ratios will be 9:7 black to colorless.
In this case, normal is often called Z+ or X+,
since it is able to make Z and/or break down X, while mutant is called Z- or X-
since it is unable to make Z and/or breakdown X. Using this terminology,
phenotypic ratios will be 9:7 Z+: Z-.
2. What if the 2 genes affect different steps in the same pathway?
Now suppose the 2 peptides (products of genes A and B) catalyze different steps in the same pathway. For example, suppose
a. Z+ vs Z-. (Inactive gene A or inactive gene B gives the same phenotype)
Phenotype:
If
you define normal phenotype as ability to make Z, then defect in gene/enzyme A or
gene/enzyme B produces the same effect = a block in the pathway. (Although a different
intermediate will build up in each case.)
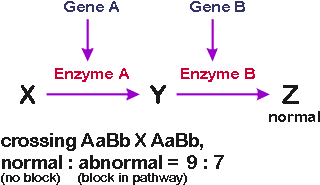
Ratios: If you are only interested in ability to make Z,
a cross of AaBb X AaBb gives normal: mutant phenotype in a ratio of 9:7. (Lack
of product of gene A has same phenotype as lack of product of gene B, & so ratios
are same as in case discussed above).
What if Z is colored? If both X and Y are colorless (but Z
is pigmented), then ratios will be 9:7 of pigmented (normal) to colorless
(albino).
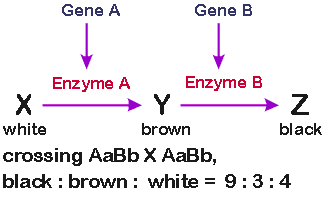
b. Epistasis. (Inactive gene A gives a different phenotype than inactive gene B)
Phenotype: Suppose X
and Y serve different functions (other than to make Z) and/or X, Y and Z are different
colors (as indicated below). Then a block in X →Y looks/acts differently than a block in
Y → Z.
For example, in the case shown below:
Ratios: In this case, if you define phenotypes by color, a cross of AaBb X AaBb → offspring with three phenotypes in ratios of 9 (black): 3 (brown) :4 (white).
This
type of situation is
known as epistasis -- effects of gene A (when it is aa in this case) override (are epistatic to) the
effects of gene B. In other words, if genotype is aa, it doesn't matter what the state of
the B's are -- aa__ is always white. The gene controlling the first step is said to be
epistatic to the gene controlling the second step.
For a similar case, with a
picture, see Sadava fig. 12.14 (10.14).
c. Epistasis vs Dominance. Epistasis is sometimes confused with dominance, as both cases refer to how the effects of one allele or gene overrides the effects of another. However they are quite different.
(1). Dominance refers to the effects of one allele overriding the effects of another allele (of the same gene). For example, A is dominant to a.
(2). Epistasis refers to the effects of one gene overriding the effects of another gene. For example, gene A (really the aa genotype) is epistatic to gene B.
d. General Case.
See problem set 10 for examples and to test your understanding.
Note: In the 3 previous examples, the genes were called gene A and gene B, and the alleles were A, a, B & b. It is probably a better practice to call the genes and the alleles by different names to avoid confusion. In the following example, the genes are called alpha and beta to keep them straight from the alleles A and B.
To get practice in matching up patterns of inheritance and the underlying metabolic pathways, try 10-4, 10-15 & 10R-5.
IV. Introduction to Linkage and crossing over -- Suppose gene alpha (alleles A and a) & gene Beta (alleles B and b) are on the same chromosome. What are the genetic consequences?
A. What gametes do you get? (Left Panel of Handout 22B)
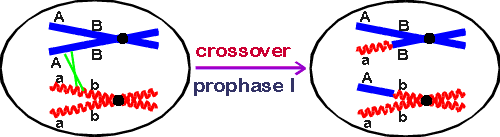
Suppose two genes, alpha and beta, are on the same pair of homologs. In any particular meiosis, there can either be no crossover in the region between between the 2 genes (arrow (1) on handout, left panel) or there can be crossovers in the region between the 2 genes. The results of a single crossover are shown on the handout (arrow (2) on left panel); also see picture below.
1. If there is no crossover between genes alpha and beta, what gametes will you get? Only 2 kinds, AB and ab, in equal proportions. (This is assuming that you start with AB on one homolog and ab on the other.)
2. What if one crossover occurs between the two genes? In that case, the equivalent parts of two homologous (not sister) chromatids are exchanged (see handout) switching an A and an a allele. What gametes will you get this time? The result will be 4 different gametes, two that are AB and ab (the parental combination), and two that are Ab and aB (called recombinants). See Sadava fig. 12.19 (10.19) or Becker fig.20-16 (20-17).
B.
Terminology:
1. Parents. If you have a double heterozygote, there are 2
possibilities: you can start with
(1) AB/ab or (2) Ab/aB. Letters before slash = alleles on one chromosome (from one parent);
alleles after slash = alleles on other homologous chromosome (from other parent).
This way of writing the genotype of a diploid is used because it is easy to
type. Although it is the standard way of writing genotypes, it is often easier
to understand what is happening if you draw out the two homologs and write the
alleles in the corresponding positions (loci) on each homolog. Here is an
example:
| (1) | (2) | |
| AB/ab | Ab/aB | |
| ----A--------B---- | ----A--------b---- | |
| ----a---------b---- | ----a--------B---- |
2. Products of Meiosis. Products of meiosis (gametes or spores) can be classified as parental or recombinant
Parental = has alleles that were on one homolog in the heterozygous parent = combination of alleles you started with (before meiosis).
Recombinant = has a new combination of alleles on one homolog.
| Diploid Parent | Parental Gametes/Spores | Recombinant Gametes/Spores |
|
AB/ab (1 above) |
AB and ab | Ab and aB. |
| Ab/aB (2 above) |
Ab and aB | ab and AB |
3. Linkage -- See the left panel of Handout 22B.
a. Definition: Genes are said to be linked if there are more parental gametes than recombinant ones. Linkage is said to be zero when the number of recombinants and parentals is equal (more details next time).
b. Mechanism: If the two genes of interest, in this case genes alpha and beta, are relatively close together, most meioses will occur without a crossover between the genes, and most gametes will be the parental type. (In this case, most meioses follow arrow (1) on the left panel of 22B.) However there may be some recombinant type gametes from the meioses with a crossover. (These are meioses that follow arrow (2).)
c.
Linkage and Distance: The number of recombinant gametes and the
number of meioses that follow arrow (2) will increase as the
distance between alpha and beta increases (up to a point)*. As distance & the number of
recombinants increases, linkage decreases. How linkage and distance are treated
numerically will be discussed later.
d. Multiple crossovers:
For now we are ignoring multiple considers -- we are assuming there is only
0 or 1 crossover per meiosis. The effects of multiple crossovers will be
considered next time.
C. What is the significance of all this? Why does linkage and crossing over matter?
1. Biological significance. Crossing over allows more combos by reshuffling existing alleles and/or mutations. Without crossing over, the same alleles would remain together on each chromosome (homolog) indefinitely. Allowing more combos provides more raw material for natural selection/evolution. Crossing over also allows repair -- if a and b are defective alleles, crossing over allows you to get a chromosome with both A and B from Ab plus aB.
Note: In eukaryotes, crossing over occurs only at prophase I of meiosis. It doesn't occur at mitosis. Therefore, crossing over only affects the next generation, not the generation in which it occurs. If crossing over occurs in the germ cells of a multi-cellular organism, the gametes of the organism are changed, but the somatic cells of the organism are unaffected.
2. Laboratory and/or medical significance. The frequency of crossing over between genes provides positional information that allows the making of maps, and linkage can be exploited for predictive purposes and other applications, as explained below.
To review the expected gametes and terminology, see problem 10-6 esp. B-C. (More details on terminology are below in VI.)
V. An example of the use of linkage -- How the HD gene (the gene that causes Huntington's Disease when defective) was located and cloned.
A. Without a marker (linked gene) -- Who will get HD?
1. The Problem. Symptoms of HD don't develop until late adulthood (usually). How to tell who will get the disease?
Disease is dominant; causes degeneration of nervous system. (H allele causes disease; h is the normal allele that does NOT cause disease.) What gene or protein involved was unknown (until recently, when gene isolated). For more background, see the 'First Mention' column of the NY Times, from 12/8/09. The column describes the first time HD was discussed in the newspaper.
2. Prediction (pre-linkage). Suppose there is an affected parent and normal parent. They have a kid. What is chance kid will have HD? (You figure it out. Is affected parent likely to be Hh or HH?) Without any DNA testing, kid will have to wait 40-50 yrs to find out if s/he has disease or not, well after deciding whether or not to have kids. This is not optimal!
B. With a marker -- Who will get Hd?
1. The Idea. Suppose you have a parent who is AB/ab, and genes alpha and beta are closely linked. Then if a kid inherits allele A, s/he will probably get allele B but if kid inherits the 'a' allele s/he will probably get the 'b' allele. In a similar way, if you have a gene linked to the HD locus, then you could infer which allele of HD the kid got from which allele of the linked gene was inherited. In the case of HD, and many similar diseases the "linked gene" was really not a gene but a variable section of the DNA called a polymorphism. (Actually an RFLP -- see handout 17A & below.)
2. Polymorphisms. There are regions of the DNA that are very variable. These regions are said to be polymorphic because they come in multiple forms (with different base sequences). These highly variable regions often occur in sections of the DNA that do not affect phenotype -- in spacers, introns etc. These various forms or polymorphisms are important (even though they have no effect in phenotype) because the variations can be used as the basis of identifications, as previously explained. These differences can also be used as "genetic markers" that can be followed through crosses. You can follow the multiple forms of the DNA sequence through a pedigree just as you would alleles A vs a or B vs b.
3. Haplotypes. A polymorphic section of the DNA comes in multiple versions that are distinguished by their differences in the number and/or lengths of fragments generated by PCR, restriction endonuclease digestion, etc. Each version is often called a haplotype. A haplotype is similar to an allele in that it is one alternate version of a DNA locus. The term "allele" is usually used to refer to a section of the DNA that has a known function; differences in alleles are reflected in differences in phenotype. The term "haplotype" is usually used to refer to a section of the DNA that has no known function (or whose function is irrelevant). Different haplotypes are distinguished by their differences in DNA sequence. The "phenotype" is the genotype = the state of the DNA. Just as different alleles of the same gene can be be followed through crosses, so can different haplotypes.
4. The details for HD (These may not all be covered in class.)
a. The polymorphism. The gene that causes HD (when defective) is linked to a polymorphic region with 4 alternatives, or haplotypes, known as A, B, C & D. These haplotypes are distinguished by how they are cut by a particular restriction enzyme. In the DNA near the HD gene, there are 4 restriction sites, of which two are invariant and two are polymorphic (variable).
Short Version:
Sites: With respect to the polymorphic
sites, a DNA molecule can be - - (A) , + + (B) , + - (C) , or -+ (D). The '+'
means there is a sequence recognized and cut by the enzyme; a '-' means the
sequence is not recognized by the enzyme.
Haplotypes: The haplotype is determined by
digesting the DNA and doing a Southern, using a probe to the region between the
two invariant sites. (See table below.)
Probe: How did they get the probe to the polymorphic region
linked to HD? By brute force = hard work + luck. They identified many
polymorphic regions in the DNA, and made probes to them. Then they analyzed the
pattern of inheritance of HD and the pattern of inheritance of the various polymorphisms
(using the probes). They kept at it until they found a polymorphism inherited
along with HD.
Long Version (for reference): The DNA looks like this:
| ↓ | (1) | (2) | ↓ | ||||
|
<------------10kb------------> |
<-------4kb-------> |
<---------8Kb---------> |
H or h allele |
Sites marked with a vertical arrow are not polymorphic; (1) & (2) are polymorphic sites. In some individuals (1) is mutated and so is not cut by the restriction enzyme; in others, (2) is mutated and in some people, both are mutated. Any individual DNA molecule/chromatid/chromosome can be cut at one or both of the variable sites, so there are 4 versions (haplotypes) of this region:
| Haplotype | Site (1) Cut? | Site (2) Cut? | Size of pieces | # of pieces |
| A | - | - | 22kb | 1 |
| B | + | + | 10, 4, 8 kb | 3 |
| C | + | - | 10 & 12 kb | 2 |
| D | - | + | 14 & 8 | 2 |
People have 2 chromosomes, so they have a mixture of pieces that depends on the sites on both chromosomes, but from the set of pieces you can tell if a person is AA or AB etc. See below and problem 10-17 for an example of how to analyze a family tree using these RFLP's. (Note that the problem refers to the marker region as the A/B/C/D gene.)
b. Role of the RFLP A linked polymorphism can be used as a "marker" to indicate which chromosome the child has inherited from the affected parent, as in the example below.
c. An example: Suppose normal parent is AA and affected parent is AB and H, and you know (from family history) it's A H/B h (H allele on same chromosome as A). If kid is AB, will s/he be likely to get HD? If kid is AB, and there was no cross over, kid got chromosome from affected parent with B & h and kid will be okay. If kid is AA, kid will get H (if there was no crossover). Note that you need a family history to know whether affected parent is A H/ B h or A h/ B H.
d. Additional Details
(1). A test like the one described above (using linkage between a polymorphic region and the HD gene) was used for
predicting inheritance of HD until the gene itself was isolated. (Now you can test for
the sequence
of the gene itself.) Many other similar
procedures have since been developed for predicting inheritance of other genetic
diseases.
(2). Phase. Does HD (or whatever disease we are talking about) always go with same particular haplotype of the A/B/C/D region (or whatever the linked marker gene is)? No. In any particular family, HD can go with any version of the A/B/C/D region. It depends which haplotype (A, B, C or D) was on the chromosome at the time the original mutation of h → H occurred.
(3). Effects of crossing over. The closer the linked marker (such as A/B/C/D) is to the disease gene, the more accurate the predictions can be, because the lower the chance of crossing over between the "marker" and the actual disease gene.
To review linkage of HD to the A/B/C/D locus, see problem 10-17. For additional examples of using polymorphisms as "markers" to follow in crosses, see 10-18 (A-D), and 13R-4 to 13R-6. (13R-7 is a good review.)
C. Positional Cloning. Problems of phase and crossing over (see above) can be eliminated by identifying the gene itself. How to do that? The HD gene and many other disease genes have been located by their linkage to a polymorphism. The genes were cloned (see below) on the basis of this "positional information" -- their position relative to some variable site.
1. How do you find a gene responsible for
a hereditary disease?
In the "old days" scientists would find out what
protein was missing/abnormal and use this info to pin down the gene responsible.
(From knowledge about the protein you'd get the mRNA and make a cDNA probe, or
make an oligonucleotide probe, etc. Then you'd use the probe to find the gene.) The
standard approach was to go from protein to gene. This was considered "forward"
or standard genetics. (It's backwards relative to central dogma, but the classical way of doing
it.)
Nowadays, scientists have done all the cases that can be easily
done this way. Now we have lots of inherited diseases where the protein that is
defective is unknown. What to do? You find a gene or marker (spot in the DNA) linked to
the disease gene (by pattern of inheritance) and use linkage = position = to find the gene
(as explained further below). Then you sequence and decode the gene to see
what protein it makes. This is "reverse" genetics or positional cloning; first
you find the gene (by what it is linked to) and then you figure out what protein the gene
codes for (from the DNA sequence).
2. Why examining and/or isolating a gene itself is better than
following a linked marker
a. No need to worry about crossovers between
the marker and the allele that causes disease.
b. No need to worry about phase -- don't have
to figure out which allele of marker (really which haplotype) goes with the disease allele
in that particular family.
c. From gene sequence, can get protein sequence
and hopefully get hints on treatment and/or do gene therapy. This has not led to much
practical treatment so far, but many methods of gene therapy are currently in the works.
3. Chromosome walking -- How gene is isolated once its position
is known (we'll discuss idea/outline only, no details).
Once you know area where gene is (near some polymorphism) you isolate (clone)
regions of DNA nearby. It's called chromosome walking because you look at successive
overlapping sections of the DNA as you go down the chromosome, away from the
polymorphic site (usually
in both directions). * You keep looking at successive sections of DNA until you find one
section that is always messed up in diseased/affected individuals. (Messed up here means
clearly changed -- can 't be properly spliced, or translated, etc.) That section must be
the gene responsible for the disease in question.
* You cut up the DNA in a way that gives you a library (or libraries) containing overlapping fragments. You use a probe made of the end of one fragment to fish out the next, overlapping fragment. For a picture see here.
Next time: The details of linkage & crossing over; then how you analyze the genetics of populations, not just individuals.
© Copyright 2010
Deborah Mowshowitz and Lawrence Chasin. Department of Biological Sciences
Columbia University New York, NY
Last Updated: 12/06/10 03:56 PM