{kind=link}
C2005/F2401 '07 Lecture #12 -- Wrap Up of DNA Synthesis; PCR; What is RNA & What is it Good For? Next time: How is RNA Made?
Handouts: 11-3 -- DNA Replication - Details at Fork & 12A -- PCR (Not on web.) 12B = code table and ribosome structure. (See texts.)
© Copyright 2007 Deborah Mowshowitz and Lawrence Chasin,
Department of Biological Sciences, Columbia University New York, NY. Last updated
10/16/2007 09:49 AM
References are to 6th ed. of Becker and 8th ed.
of Sadava. (Refs in parentheses are to previous editions, 5th for Becker &
7th and 6th
for Purves/Sadava.)
For nice animations of the Meselson-Stahl experiment and DNA replication (including primers) go to
http://highered.mcgraw-hill.com/sites/0072437316/student_view0/chapter14/animations.html#
These animations (& your texts) may mention enzymes not discussed in class; you are not responsible for the extra details. A box with the animation should appear on the upper left when you click on the appropriate link. If nothing happens when you click on the link, the box may be behind the page you are looking at. Shrink the main page to see the animation.
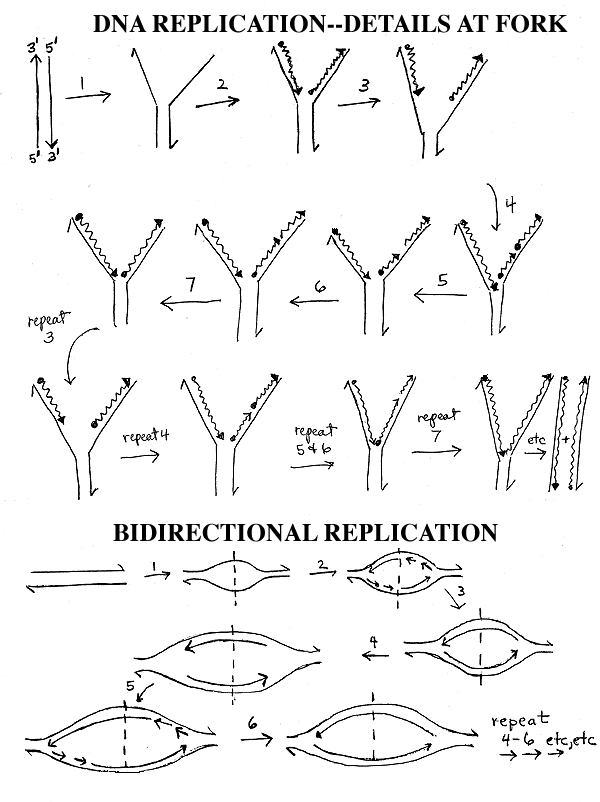
I. Discontinuous synthesis, cont. Topology of Forks
A. What Moves? In the pictures on the handouts and in many pictures in the texts, it looks like the DNA stays put and the enzymes move down the DNA. It is probably the other way around -- the enzymes stay put, and the DNA slides through them. See Sadava fig. 11.13 & 11.14 (11.11 & 11.12 in 7 th ed, 11.12 & 11.13 in 6th; not in 5th edition.)
B. How Polymerase is Oriented: (FYI only.) The polymerases making the leading and lagging strands are probably moving (or facing) effectively in the same direction, relative to the fork, because the template for the lagging strand is looped around. See Becker fig. 19-14. (The leading and lagging strands are both being made in the 5' to 3' direction, anti-parallel to their respective templates.) The end of the animation (see link above) called 'How nucleotides are added in DNA Replication' shows the looping.
II. Primers & Primase. (Top of
handout 11-3. Steps 5 & 6)
A. The Problem
If you put DNA polymerase, ligase, pyrophosphatase,
dATP, dGTP, dTTP & dCTP in a test tube (+ all unwinding enzymes) will you get DNA? No, because DNA polymerase can't start a new chain
-- it can only add on to the 3' end of a pre-existing chain. So how do new
DNA strands get started? Using primer and primase.
B. The Solution in vivo
1. How Primase makes Primer -- see
Sadava 11-16 (11-14 in 7th ed, 11-15 in
6th ed) or
Becker 19-11 (17-11).
a. Primase: Primase is a type of RNA
polymerase that uses nucleotide triphosphates to make a short RNA stretch (probably less
than 20 bases long {Q&A}) complementary to the 3' end of the template (= 5'
end of new strand). RNA polymerases (unlike DNA polymerases) can start new
chains from scratch.
RNA chains are made 5' to 3' in much
the same way as DNA, using ribo-nucleoside triphosphates (containing U, not T) instead of deoxy-nucleoside
triphosphates.
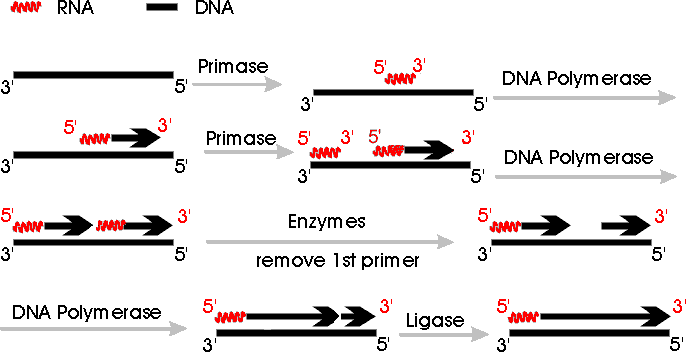
b. Primer: Short RNA stretch made by primase is called primer. On handout (11-3) of events at fork, RNA primer is represented by a dot. (In diagram below, primer is a red squiggly line.) Primase catalyzes synthesis of primer, and then DNA polymerase adds on the 3' end of the RNA primer.
2. How
Primer is Removed & Replaced
The primer (the short RNA section) must be
removed and replaced by DNA. The process is shown in steps 5 & 6 of handout
11-3 and in the diagram below. Some of the steps below may occur simultaneously,
but are described separately to make the process
clearer.
Step 5: Primer is removed, leaving a gap between the 3' end of Okazaki fragment #2 and the 5' end of fragment #1, giving molecule E.
Step 6: DNA polymerase adds on to the 3' end of primer #2 to fill the gap, giving molecule F.
Step 7: Ligase joins the loose ends of the lagging strand, giving molecule G.
Removal of RNA primer (step 5) and filling of the gap with DNA (step 6) may occur at the same time, using two different catalytic parts of a single enzyme.
3. Summary Pictures of Use & Replacement of Primer
See Becker Fig 19-13 (17-13) or Sadava 11.19 (11.17 in 7th ed, 11.18 in 6th ed) or Picture Below. Note:
Some of the pictures in the older editions of the texts don't have all the details right.
Some of the figures imply that DNA can replace RNA primer
without the need for a free 3' end for DNA polymerase to add on to. Other
figures show ligase joining the Okazaki fragments at the wrong place. (See
picture below or solution to problem 6-14, part B-3, for correct position of
ligation. Note that the replication fork in problem 6-14 goes in the opposite
direction from the fork in the picture below.)
In the picture below, which summarizes the process of primer synthesis and replacement, all arrows go 5' to 3'. Only one side of the replicating fork is shown -- the side carrying out synthesis of the lagging strand. The side carrying out continuous synthesis is omitted. Note that replication fork below goes right to left -- DNA is unzipping from right to left.
Function & Replacement of Primer; see also handout 11-3.
For animations of primer removal and other events at the replication fork, see the links given above at the start of the lecture.
C. Telomeres and Telomerase -- a
Biological consequence of the need for primers
1. The "loose end" Problem
There is no easy way to replace the primer on the left end of the new strand (in picture above); also see Becker fig. 19-15 (17-14). The RNA can be removed, but no DNA can be made to fill in the gap.
2. Solutions
a. Small DNA's
(& most prokaryotic chromosomes) are generally circular, which circumvents
this problem.
b.
Linear chromosomes (the norm in eukaryotes) tend to get shorter with each
replication -- in next go round, chain that has just been made will be
template, and it is shorter than the original by the length of the primer. The
consequences will be discussed next term when we focus on eukaryotes.
FYI: Eukaryotic chromosomes get shorter with each replication, but it doesn't usually matter because the DNA molecules in the chromosomes have special repeated structures called telomeres on the ends. The repeats are gradually lost unless replaced by an enzyme called telomerase. See Sadava 11.21 (11.18 in 7th ed, 14.3 in 6th) or Becker 19-16 (17-15). (Note: in Purves' picture in the 16th edition, the wrong strand is "too short." The 3' end should be longer than the 5' end, not the reverse.) The lack of telomerase may be what limits normal somatic cells to a finite life span of 50-60 divisions; the presence of telomerase may be what allows indefinite multiplication of cancer cells. Germ cells, that produce eggs and sperm, make telomerase, so a new generation always starts out with full length telomeres. For an animation of how telomerase works, see http://faculty.plattsburgh.edu/donald.slish/Telomerase.html. (Note that this is for eukaryotes; in this case there are two different DNA polymerase for the leading and lagging strands. Both grow chains in the 5' to 3' direction.)
To review primers, see problem 6-12, A-D and problem 6-14. If you have the 2004 ed. of the problem book, there is a typo in problem 6-12, part D. The later editions are corrected.
Reminder: In the latest edition, the
answers to problem set 6 are on Courseworks; not in the problem book. We
apologize for the mix up.
For all
corrections in notes, current and previous editions of the problem book, etc., see the
corrections page.
IV. PCR (Polymerase Chain
Reaction) -- Handout 12A. A Practical Application of the need for Primers.
The inventor, Kary Mullis, received the Nobel prize in 1993. For his acceptance speech, biography, etc. see the Nobel Prize official site. Also see class handout.
A. Idea of prefab primer, hybridization.
DNA synthesis will not start without a primer. In a living cell, primase (a type of RNA polymerase) makes the necessary RNA primer. Then DNA polymerase can take over, adding on to the 3' end of the primer. In a test tube, you can omit primase and use an oligonucleotide (short polynucleotide, usually DNA) as primer (= prefab DNA primer) to force replication to begin wherever you want. The primer you add will hybridize to its complementary sequence, wherever that happens to be (not necessarily at the end of the DNA) and DNA polymerase will add on to the 3' end of the primer, thereby starting elongation of a chain from wherever the primer is.
B. Steps of PCR -- see PCR handout (12A), Sadava 11.23 (11.20 in 7th ed, 11.21 in 6th), and/or Becker Box 19 A (17A). For an animation, go to http://www.dnalc.org/ddnalc/resources/pcr.html
The site listed above (The Dolan DNA Learning Center) has many good features you may want to check out. There is a list of additional animations on PCR, DNA replication, etc. at http://www.dna.utah.edu/PCR_Animation_Links.htm. Please let Dr. M know if you find any of these sites (or any others) particularly useful.
1. First Cycle: You denature your template (A) and add primers (one to each strand) to the denatured DNA (B). Then you cool the mix down, and each oligonucleotide primer hybridizes to its complement (step 1 on handout; gives D). The two long strands of template do not renature. Then DNA polymerase adds on to 3' end of primer until it reaches the end of the template strand (step 2 = elongation). This completes the first cycle (ends at E). The new strands you just made (dashed on handout in E) include the target sequence, plus some extra DNA on their 3' ends. (This "extra" corresponds to the sequence between the target area and the 5' end of the template strand.)
2. Second Cycle: You heat the DNA to denature it (step 3), and add more of the same primers as before (step 4). Then you allow DNA polymerase to add on to the 3' ends of the primers (step 5). This completes the second cycle (ends at H). On the handout, only the fate of the new strands made in cycle two is shown after steps 4 & 5. The old strands simultaneously go through another cycle just like the one above (steps 1 & 2), but this is not shown on the handout. The new strands you made in cycle 2 (shorter strand of each molecule of H) include only the target sequence.
3. Third Cycle: You heat the DNA again to denature it (step 6), add primers (step 7) and allow DNA polymerase to add to the primers (step 8). This completes the 3rd cycle (ends at K). On the handout, only the fate of the new strands made in cycle two is shown after steps 7 & 8. (The fate of the complementary strands, left over from the previous cycle, is to repeat steps 4 & 5.) At the end of this cycle, you finally have double-stranded DNA molecules the length of the target sequence (see K).
4. Additional Cycles: After each cycle you heat the reaction mixture to denature the DNA, and then you cool the mixture down to start the next cycle. In each cycle, primer sticks to the appropriate spot (its complement) and polymerase starts at the 3' end of the primer and goes to the end of the template. Note that primers are complementary to sequences in the middle of the original chain, but that after two cycles the parts beyond the primers are no longer copied. {Q&A}.
5. How reaction is actually carried out. All components (template and excess of heat resistant polymerase, primers & dXTP's ) are present from the very beginning. The mixture is heated and cooled repeatedly to end and start subsequent cycles. You don't have to add primers, polymerase, etc. to start each cycle.
6. How many Primers? New molecules of primer are used in each round. However, the primer molecules used in each round have the same sequences as the ones used in all the previous round. The primers are not reused -- new primers (with the same sequences as before) are needed for each cycle. You need only two types (sequences) of primer, but you need many molecules of each.
7. Identification of Product. The products of the PCR reaction are usually identified by their lengths, which are determined by gel electrophoresis. Gels are used that separate DNA molecules on the basis of their molecular weights (which depends on chain length). Hybridization to labeled probes is often used to detect the positions of the bands of DNA on the gel. (More on this later.) An animation of DNA gel electrophoresis is at http://www.dnalc.org/ddnalc/resources/electrophoresis.html.
C. Special Features of PCR (as vs. regular DNA synthesis)
1. Special Polymerase. The DNA polymerase used in this procedure is a special heat-resistant one (called Taq polymerase) that is not denatured when the temperature is raised to separate the two strands of the DNA. This special polymerase was isolated from bacteria that live in a hot spring.
2. No discontinuous synthesis. Note that the entire template molecule is denatured completely before each cycle, so each strand can be copied continuously. There is no replication fork and thus no discontinuous synthesis here.
3. Preformed DNA primer. Primase is absent, so no RNA primers are made. Oligonucleotides of DNA (not RNA) are added instead to act as primers.
To review the
PCR technique, see problem 6-13, C-1 and 6-15.
For an animation of PCR and links to animations of other DNA techniques, see the
url listed above under B.
D. Uses/Advantages of PCR
1. Amplification: Uses small number of starting molecules & produces large number of copies of target sequence.
The beauty of this scheme (PCR) is that the desired (target) sequence is copied exponentially and the other parts of the original DNA are copied linearly. So after a few cycles you have lots of copies of the target sequence (and not much of anything else). To convince yourself of this, see the answer to problem 6-13, part C-2. To use this technique and make many copies of the target sequence all you need (in theory) is ONE starting DNA molecule (and appropriate primers). Given current technology, you need 10-50 starting DNA molecules (see articles handed out in class). You can use the multiple copies for many different purposes such as characterization and/or identification as explained below.
2. Detection -- Can be Used to see if a particular target DNA is present or not.
You can add primers to a sample that you suspect contains some particular target DNA, such as HIV DNA, or DNA from genetically modified corn, or DNA from a species of protected whales. The primers are complementary to a sequence found only in the target DNA -- the one you are testing for. (In the cases mentioned, the primers would be complementary to a sequence in HIV DNA, or a sequence added to ordinary corn DNA by genetic engineering methods to make the special corn, or to a DNA sequence unique to the protected species of whales.) Then you see if polymerase can make DNA. If no target DNA is present, primers will have nothing to hybridize to, so polymerase will have nothing to add on to, and no copies of DNA will be made. So if you don't get multiple copies, it indicates there was nothing to copy -- your target DNA was not there. If you do get multiple copies, your target DNA was in the sample.
Notes: (1) The standard HIV screening test is not for HIV itself or for HIV DNA but for antibodies to proteins of HIV. (PCR is used as a backup to confirm a positive result with the standard screening test, or to measure the actual levels of HIV.)
(2). Why would you test for genetically modified corn? StarLink corn is a type of genetically modified corn that was approved for animal feed, but not for human use. In spite of attempts to keep it separate, it has turned up in many human foods. It is probably harmless to humans, but no one wants to take any chances. Testing for the modified DNA is the only way to tell if StarLink corn (or any other genetically modified food) is present in a mixture or not. A site with an explanation of the StarLink fiasco is at http://www.geo-pie.cornell.edu/issues/starlink.html.
3. Forensics -- Can be Used for identification -- DNA fingerprinting
a. Basic idea: PCR can be used to copy specific sections of the DNA from different samples -- for example, from DNA left at the scene of a crime and from DNA from a suspect. The sections of amplified DNA can then be compared to see if they match or not (in length, sequence, etc.). The sections that are compared are highly variable ones that probably don't carry any information and are merely spacers in the DNA. If enough sections are checked, you can determine (to a very high degree of certainty) whether the two DNA samples came from the same person or not. DNA testing can be used to identify the guilty (inclusions) and to clear the innocent (exclusions). Alec Jeffreys, who first came up with the idea of using DNA testing for identifications, received a Lasker award in '05. For a pdf with details see the Lasker site.
b. Examples: See articles handed out in class and article from the San Francisco Chronicle of 10/19/99. (Note: you'll need to go to the SFChronicle web site itself if you want to see the pictures or get some of the older articles.)
c. Inclusions: If the samples match at enough highly variable spots, then there is a very high probability the samples came from the same person, because the degree of variation is so high that only a few different people in the world should have the same pattern.
d. Exclusions: If the two samples do not match, then it is clear that the two samples came from different individuals and the suspect could not have committed the crime (since the DNA at the scene came from someone else).
e. STR's: The variable sections that are tested are often ones that have different numbers of short tandem repeats (STR's). The primers hybridize to regions outside the section with the repeats. The number of repeats in each DNA can be figured out from the length of the sections amplified by PCR. The new FBI data base contains the information from checking 13 sections with variable numbers of STR's.
For a great site from the Dolan Learning Center with examples of how DNA is used for identification and forensics click here.
4. Why you can't do this with proteins
There are very sensitive tests for presence of proteins (usually using the catalytic activities of enzymes and/or binding abilities of antibodies), but no way to amplify (make copies of) what you detect. You can't make more. PCR takes advantage of fact that DNA replicates for a living to make more copies.
Note: So-called DNA fingerprints are characteristic of the person/DNA from which they came. So-called protein fingerprints are characteristic of the protein from which they came. That's why both are called 'fingerprints.' However the two types of 'fingerprints' are made differently and used for different purposes.
V . Central Dogma -- How does DNA do job # 1?
A. Big Picture. So we have a big DNA that includes a particular gene = stretch of DNA coding for a single peptide; how will we make the corresponding peptide?
Note: gene usually means a stretch of DNA encoding 1 polypeptide, but there are complications as we'll see later.
1. Basic idea -- see also Becker fig. 21-1 [19-1] or Sadava 12.2 & 12.3:
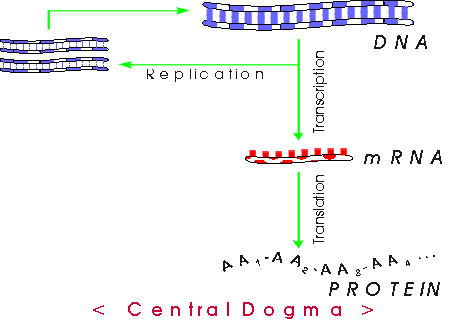
2. Terminology:
Replication = DNA synthesis using a DNA template.
Transcription = RNA synthesis using a DNA template.
Translation (usually) = protein synthesis using an RNA template. In some contexts, translation can mean the entire process of DNA → RNA → protein.
B. What RNA is
1. Structure: See Sadava fig. 3.24 (3.25 in 7th ed, 3.17 in 6th) and table 3.3 or Becker table 3-5 (3-4) & fig. 3-17 for comparison of DNA and RNA. RNA is single stranded (although sections may double back on themselves → double stranded regions), has U not T, ribose not deoxy and is generally shorter, but otherwise like DNA. RNA is less stable than DNA -- more easily damaged (because of reactive OH on ribose and because a single strand is more exposed) and less easily repaired (because no 2nd strand to use to correct mistakes on first strand). DNA is also more easily repaired because it has T not U, so damaged C's (which are oxidized to U) can be recognized and removed. In summary:
DNA RNA Significance/Effect of Difference Double Stranded
Single Stranded*
For RNA: Ease of repair down; likelihood of damage up.
T not U
U not T
For DNA: Ease of repair of damaged (oxidized) C up. (Damage that coverts C to U can be detected & repaired.)
Deoxyribose
Ribose
For RNA: Reactivity up, stability down
Very long
Relatively Short
For RNA: Less Information carried per molecule but molecule is much more convenient size
* RNA is basically single stranded, but can fold back on itself to form hairpins -- short regions that are double stranded. See Sadava 3.25 (3.26 in 7th ed.)
2. Synthesis. RNA grows just like DNA by adding nucleoside triphosphates (XTP's) to the 3' end of a growing chain. For RNA, enzyme for elongation is called RNA polymerase, XTP's are ribo (not deoxy) and U replaces T. Details to follow.
3. Types. There are 3 major types of RNA involved in translation: messenger RNA (mRNA), transfer RNA (tRNA) and ribosomal RNA (rRNA). The roles of the different types of RNA are explained below.
VI. Why mRNA?
A. Basic idea: mRNA = Working, disposable copy vs DNA = archival, permanent master copy. DNA = big fat comprehensive reference book or complex web site. mRNA = Xerox of one (book) page or print out of one web page with information you need for a particular assignment. Book stays safe in library; web site remains unchanged. Xerox goes to your room, is actually used, gets covered with coffee stains, smudged, and thrown away.
B. How function of mRNA corresponds to structure
1. Convenience -- small size (1 or a few peptides' worth) is much more convenient than many genes' worth. Xerox of one page much more convenient to work with than big fat book.
2 . Flexibility in different rates of synthesis of different proteins. Cells can vary the amount of mRNA made/gene so they can vary the rates for synthesis of different proteins . If mRNA is template for protein synthesis, a cell can make many mRNA copies of DNA template for protein B if it needs to make lots of protein B. If DNA were direct template, cell would have only 1 copy of template no matter how much protein B it needs to make. (If several people are working on the same assignment, you need more Xeroxes.)
3. Flexibility in response to needs that change with time -- bacterial cells constantly make new mRNA's, and degrade old ones (to mononucleotides) so a cell can constantly adjust the rates of protein synthesis (and change which ones are made) to meet the current need. DNA has a long half life, as expected of a master copy ; bacterial mRNA has a short half life, as expected for a working copy. (When assignment is done, you crumple up the Xerox and throw it away. Then you make a new Xerox for the next assignment.) See Becker, Key Aspects of mRNA metabolism, p. 680 (654) at end of Ch. 21 (19).
Note: In eukaryotes the situation is more complex -- some eukaryotic mRNA's last a long time while some are short lived. It depends on the cell type and what protein the mRNA codes for.4. Preserve Master. Using mRNA to make protein saves wear and tear on master -- no coffee stains on the archival copy (DNA).
VII. Why other kinds of RNA
for Translation? What are tRNA and rRNA for?
A. How decode mRNA (general idea)
1. It's read in triplets . Reading starts at a fixed point and then mRNA is read one triplet or codon at a time in the 5' to 3' direction.
2. Code table (see texts or handout) lists codons = triplets found in the mRNA (NOT complements of codons) and corresponding amino acids. One codon specifies one amino acid. For example, CUA means leucine; UUU means phenylalanine, AUG means methionine.
B. What is tRNA & what is it for?
1. Need an adapter -- how does cell know AUG is met and CUA is leu? You have the text or handout with the code table, but cell doesn't.
a. Transfer RNA (tRNA) = adaptor. Cell uses tRNA to match the codon in the mRNA (say AUG or CUA) with the corresponding amino acid (met or leu, respectively).
b. Loading Enzymes. Adaptor must carry the correct amino acid. Cell uses loading enzymes to put the correct amino acids on to their respective tRNA's. More details next time.
2. Properties of tRNA
a. Size: About 75 bases long (relatively small).
b. Many different ones. Actual number of dif. tRNA's is more than 20 (#of dif. amino acids) and less than 64 (# of dif. codons). More exact estimate of # of different tRNA's to follow in next lecture.
c. Two headed molecule: tRNA has 2 ends (in its 3D form) -- one complementary to codon (= anticodon) and one (on 3' end) to pick up the appropriate amino acid (= acceptor end) with the help of the appropriate enzyme.
d. General features of structure -- every tRNA molecule is folded back on itself to form some double stranded regions. Sequences of different tRNA's differ, but all are self complementary in certain regions. Every tRNA molecule has same basic "secondary structure" = cloverleaf; this is folded into an L shaped "tertiary" structure, which has anticodon at one end and acceptor for its amino acid at the other. (See Becker fig. 22-3 [20-3], or Sadava 12.8 (12.7 in 7th ed, 12.6 in 6th), for secondary and tertiary structures.)
Important reminder: The code table lists the codons, NOT the anticodons. The anticodon in the tRNA is the complement of the triplet shown in the table.
C. What is rRNA & what is it for? Ribosomes and rRNA
1. Function. You need something to hold tRNA (two loaded ones at a time) onto mRNA while amino acids are being hooked up and you need to provide necessary enzymes for making peptide bond etc. (How many weak bonds hold a tRNA and mRNA together?)
2. Structure. Holding of tRNA etc. is done by a structure that contains both RNA(s) and protein(s). Anything made of both is called an RNP = ribonucleoprotein or ribonucleoprotein particle. This particular RNP structure = ribosome; RNA inside it is called ribosomal RNA or rRNA. So you need rRNA as well as mRNA and tRNA for translation. For pictures of ribosome structure see Handout 12B, or Sadava 12.10 (12.9 in 7th ed, or 12.8 in 6th) and/or Becker figs. 22-1 & 22-2 [20-1 & 20-2] & table 22-1 [20-1]. )
D. Summary: How does RNA make protein?
1. "RNA makes protein" means two things:
a. Need mRNA (info goes DNA → RNA → protein) -- see Sadava 12.2
b. Need several kinds of RNA to make each protein: one mRNA, tRNA (at least 30 dif. kinds) and rRNA ( 3-4 kinds). Of course you need protein too to make protein.
2. Hardware vs. Software. rRNA and tRNA are the hardware or tools or machines; mRNA is the software or working instructions or tapes/CDs/punchcards. Cells use same old hardware and constantly changing, up to the minute, supply of new software.
Next Time: We will wrap up our
outline of how RNA makes protein, and then consider:
Where does the RNA come from? So you need lots of kinds of RNA. How do you make them? All RNA is transcribed from a DNA template. See Sadava 12.5 (12.4) or Becker fig. 21-9 & 21-11 [19-9 & 19-11]. The easiest way to go over RNA synthesis, given that we've discussed DNA synthesis at length, is to compare DNA and RNA synthesis. We'll go over this next time, and then consider how the RNA is used to make protein.
© Copyright 2007 Deborah Mowshowitz and Lawrence Chasin Department of Biological Sciences Columbia University New York, NY