C2006/F2402 '11 OUTLINE OF LECTURE #13
(c) 2011
Dr. Deborah Mowshowitz, Columbia University, New York, NY. Last update 03/02/2011 04:34 PMHandouts:
13A -- Golgi transport
models
13B --
How hydrolytic enzymes get to Lysosomes
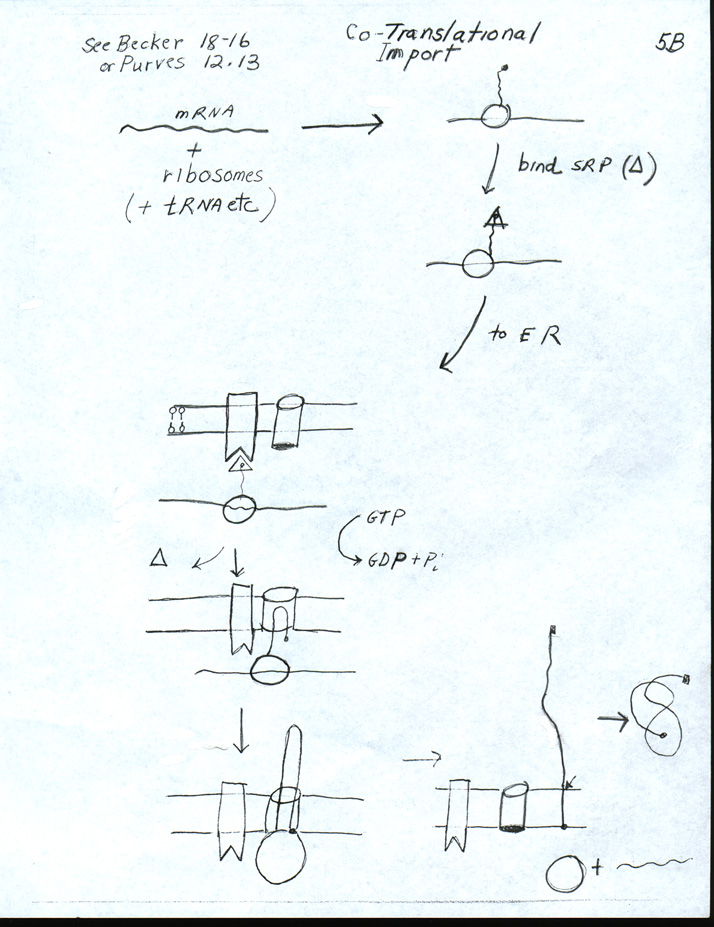
I. How does Co-translational Import Work?
(A & B = topic VII of lecture 12)
A. Signal Hypothesis -- this was covered in lecture 12 -- See Handout 12C Signal Hypothesis -- Co-translational Import
B. How do proteins cross or enter the ER membrane? See Handout 12D How proteins insert into ER membrane. and/or fig. 22-17 of Becker.
1. How proteins enter/pass through the membrane -- important points
a. SP probably forms loop not arrow. Loop enters channel (translocon) in membrane. SP loop is probably what opens (gates) the channel on the cytoplasmic side.
b. Protein enters as it is made. In humans, growing protein chains usually enter the ER as the chains are synthesized (co-translational import).
Note: In unicellular organisms, soluble proteins destined for the ER lumen often enter the ER after they are finished (post-translational import). Post translational import into the ER will be ignored here, but is covered at length in cell biology.
c. How do transmembrane proteins get anchored in the membrane? A hydrophobic sequence may trigger opening of the pore sideways, so protein slides out of pore, laterally, into lipid bilayer. These hydrophobic sequences are usually called 'stop-transfer' sequences and/or 'anchor' sequences.
d. Where will protein end up? Protein can go all the way through the membrane and end up as a soluble protein in the lumen (as in example above, on 12B) or protein can go part way through and end up as a transmembrane protein. Depends on sequence of protein.
2. Types of Proteins that can result (see handout 12D)
a. Soluble protein in lumen. Happens if protein passes all the way through the membrane and SP (on amino end) is removed, as above.
b. Integral membrane protein anchored in membrane by SP with no cytoplasmic domain. This happens if SP is on the amino end and is not removed.
c. Single Pass transmembrane protein -- get one of 2 possibilities:
(1). Type 1: Amino end is on lumen side of membrane (on E side); Carboxyl end is in cytoplasm (on P side of membrane)
One way this could happen: If SP is on amino end, and SP removed, and there is a hydrophobic sequence (acting as a stop-transfer or anchor sequence) in the middle of the peptide.(2). Type 2: Carboxyl end is on lumen side of membrane (on E side); Amino end is in cytoplasm (on P side)
One way this could happen: If SP is in the middle, not on amino end. SP in this case is not removed -- it becomes the transmembrane domain of the protein. (SP doubles as stop-transfer or anchor sequence.)d. Multipass transmembrane protein. (Requires one SP and several hydrophobic (start/stop) sequences.
(1). Hydrophobic sequences can stop the process (of moving through pore) and anchor protein in membrane, as explained above.
(2). Hydrophobic sequences in the middle of the peptide can restart looping → multipass protein. These are usually called "start-transfer" sequences (see 4).
(3). 'Start-transfer' and 'stop-transfer' sequences are probably equivalent. Role depends on where in protein they occur. (Both start- and stop-transfer sequences are also called 'topogenic sequences' as they determine the topology of the finished peptide.)
(4) A sequence that starts or restarts passage of a protein through the translocon is usually called a 'start transfer sequence' even if it also doubles as a stop or anchor in the membrane.
e. Lipid Anchored Proteins (FYI): Proteins to be anchored to lipids on the outside of the plasma membrane are generally made as follows: Protein is made on RER and inserted into the ER membrane. After the protein reaches the plasma membrane, the extracellular domain is detached from the rest of the protein and attached to lipid. (Proteins to be anchored to the plasma membrane on the inside are made on cytoplasmic ribosomes.) See Becker if you are curious about the details.
By now you should be able to do problems 3-1 to 3-3 & 3-4, A-B.
C. What is the fate of proteins made on attached ribosomes? These become part of the endomembrane system and/or leave the cell.
primarily by co-translational import as above. (There is some post-translational import, esp. in unicellular eukaryotes.) Protein can1. They enter the ER
2. Most proteins travel from ER to Golgi
3. Most proteins are sorted and processed in the Golgi
4. Where do the proteins and/or vesicles go next?
(vesicles involved in regulated secretion) → area near plasma membranea. Secretory vesicles
(1). Vesicles fuse with plasma membrane only in response to signal (such as hormone, change in local Na+ concentration, etc.)
(2). The 'signal' usually causes an increase in intracellular Ca2+, which directly triggers the fusion, causing exocytosis.
(3). Fusion results in: Release of contents outside cell
and/or addition of material to cell membrane. Click here for animation #1 -- annotated & animation #2 -- larger but not annotated.b. Default vesicles
(vesicles involved in constitutive secretion) → plasma membrane → fuse automatically (constitutively) and release contents. Same as in (a) -- leads to addition of material to membrane or outside it. HOWEVER no signal is required for fusion. This is probably the "default" for proteins that are directed to the ER but have no additional directional information.c. Vesicles containing hydrolases
→ Lysosomes (details to be discussed later).d. Vesicles containing other enzymes
→ other parts of EMS (Some enzymes may stay in trans Golgi, but others bud off and go back to other parts of Golgi, ER, etc.II. What Happens in/on the ER?
A. What happens inside ER
1. Folding of protein -- requires chaperones.
a. Chaperones (also called chaperonins) -- proteins needed to assist in protein folding. Chaperones are used every time a protein remains unfolded or becomes unfolded to cross a membrane (or refolds on the other side). Different chaperones are found in different parts of the cell.
b. Chaperones are of two major types (families) -- HSP 60 (forms barrel) or HSP 70 (binds to hydrophobic regions). Differ in molecular weight (60 K vs 70 K) and mode of action. (See an advanced text if you are curious about the mechanisms.)
c. The major chaperone inside the ER is a member of the HSP 70 family, also called "BiP"
d. Why are chaperones named HSP 60, HSP 70? HSP = heat shock protein. Chaperones, aka HSP's, are made in large amounts after exposure to high temperatures. (That's how they were first discovered.)
e. Final shape. Amino acid sequence of protein determines final, folded shape, but chaperone is needed to help reach final state.
2. Enzymatic Modifications. The appropriate enzymes inside the ER catalyze the following:
. In eukaryotes, all S-S bonds are formed in proteins inside the ER. Proteins made in the cytoplasm do not have S-S bonds. Cytoplasmic proteins do contain cysteines and have free SH groups.a. Making of S-S bonds
b. Start of N-glycosylation. Oligosaccacharides are added to the N of the amide of asparagine side chains (this is called N glycosylation.) See Becker fig. 12-7 if you are curious about the biochemical details. Additional steps of glycosylation occur in the Golgi; details below.
c. Removal of SP (sometimes)
(1). Requires signal peptidase (enzyme) and specific target sequence in substrate (newly made protein).
(2). SP is not always removed, but if it is, this is where removal occurs.
3. Some proteins stay in ER (in lumen or membrane); most move on to Golgi.
4. What happens to proteins in ER that do not fold properly? See Becker p. 750-752 (755-757) .
a. Transport to cytosol -- Unfolded proteins are transported back to the cytosol (through the translocon? another channel? -- mechanism unknown).
b. Ubiquitin addition -- in cytosol, proteins (from ER or cytosol) to be degraded are marked for destruction by addition of a multiple molecules of a small protein called ubiquitin to side chains of lysine. (See Becker or advanced texts if you are interested in the enzymatic details.)
c. Role of Proteasome = a large protein complex in cytosol that degrades ubiquitinylated proteins to fragments, at expense of ATP. Major site of degradation of intracellular proteins. (Proteins from outside are generally degraded in lysosomes.)
d. What goes to the proteasome? Proteins that are misfolded, damaged, or have served their function.
A major proportion of all proteins made in cell do not fold properly and are degraded.
Destruction of many proteins is regulated -- level of protein activity can be controlled by adjusting rate of protein degradation as well as by adjusting rate of synthesis, feed back of activity, modification, etc. More details and/or examples to follow.
e. What comes out of the proteasome? Ubiquitin (recycled) and short peptide fragments of the protein that was degraded.
f. 2004 Nobel Prize for Chemistry was awarded to Aaron Ciechanover, Avram Hershko and Irwin Rose for the discovery of the ubiquitin/proteasome system.
B. What happens on outside of ER (besides protein synthesis)
1. Lipid synthesis
eins.a. Insertion: Lipids made and inserted on cytoplasmic side (cytoplasmic leaflet) of membrane by enzymes attached to/in membrane.
b. Flipping: Enzymes ('flippases' = transporters) are required to move amphipathic lipids from one leaflet (P side) of membrane to other leaflet (E side). If lipids are moved preferentially from one side of membrane to the other, transport is active and requires ATP.
c. Transport: Lipids can reach parts of cell not connected to ER through vesicles and/or transport (exchange) prot
2. Some detoxifications and other reactions are catalyzed by proteins on the cyto side of ER. See text for details if interested.
To review the structure and function of the ER, try problem 3-13.
III. Golgi Complex -- Structure & Function
A. How things get there -- from ER in coated vesicles (coat made of protein called coatomer or COP instead of clathrin). For animation of how materials pass from ER to and through Golgi click here. B. Structure & Overall Traffic Flow - See Sadava, fig 5.10 (4.11) or Becker fig. 12-4 & 12-81. Two sides of stack
a. cis/forming face (side closest to nucleus & ER)
b. trans/maturing face (away from nucleus)
2. Three basic parts or compartments in a stack
a. CGN (cis-Golgi network) or cis Golgi -- may include fusing vesicles
b. medial cisternae (sacs) -- part in between 'cis' and 'trans' Golgi
c. TGN (trans-Golgi network) or trans Golgi -- may include budding vesicles
3. Different marker enzymes/functions found in different parts. (See Becker figs 12-5 & 12-6)
Enzymes unique to any one cell organelle or compartment are called 'marker enzymes.' = their presence is a 'marker' for the presence of that compartment or organelle.
4. Sacs in stack connected by vesicle traffic -- not completely clear which way transport vesicles go or what they carry. It is clear that newly made protein and lipid passes through the Golgi from the cis face to the trans face, as shown on this animation.
C. Function -- what reactions take place inside Golgi?
-- oligosaccharide that was added to glycoproteins in ER is modified. These oligosaccharides are attached to "N" of amide side chains of asparagines (asn's).1. Finish N glycosylation
2. Do O glycosylation of glycoproteins. Sugars are added to "O" of the hydroxyl of the side chain of ser & thr.
3. Assemble sugars of proteoglycans (linear chains of repeating sequence = GAGs)
4. Concentrate, sort proteins. This occurs at trans face (TGN). Different areas of Golgi have receptors that trap proteins going to different destinations.
To review how proteins are directed to the right place and modified in the ER and Golgi, try problem 3-4 if you haven't done it yet.
IV.
Transport
Through the Golgi -- How does
cargo (newly made proteins) move through the Golgi? Are cisternae stationary or do they progress? See handout 13A.
A. Background
1. What is known:
a. Transport vesicles
(1). Vesicles can bud off one sac (cisterna) of the Golgi and fuse with another in the same stack.
(2). In vitro, vesicles can fuse with cisternae of another stack. (See problem 3-10.)
b. Modification enzymes: Different modification enzymes are found in different parts of the Golgi (cis, medial trans). Same enzymes (marker enzymes) are always found in same part of Golgi.
c. Cargo: Cargo (newly made protein from the ER) moves through the Golgi from cis to trans.
2. Three big Issues (See table on 13A)
a. Direction of vesicle traffic: Which way do the vesicles go?
(1). cis to trans = forward = anterograde?
(2). trans to cis = backward = retrograde?
(3). Both? Older models assumed it was forward, but current evidence indicates it is both -- some vesicles go in one direction and some the other, as shown in Becker fig. 12-8.
b. What do the vesicles carry?
(1). Cargo proteins -- Newly made proteins from the ER, and/or
(2). Modification Enzymes -- Used in the Golgi to modify the newly made cargo proteins
c. Do cisternae move? What carries the newly made material from cis to trans, the cisternae or the vesicles?
d. Overall: What happens to the composition of each sac? Does the content of modification enzymes change or the cargo?
B. Models -- see handout 13A.
1. "Vesicle Transport Model" or Stationary Cisternae Model
a. Transport vesicles move primarily forward (anterograde) -- towards trans face. For an animation of this process, see here.
b. Vesicles carry cargo -- vesicles carry newly made proteins from one part of Golgi to next part for additional modifications.
c. Sacs of Golgi (& their characteristic enzymes) stay put -- newly made proteins (cargo) pass from sac to sac by means of vesicles.
d. Net Result: Enzyme composition of each sac stays the same. Each part stays in the same place and holds on to its characteristic ('marker') enzymes. It is the substrates of the enzymes (the newly made cargo proteins) that pass through, carried by the vesicles from sac to sac.
2. "Cisternal Maturation Model" = a modified Progression Model
a. Transport vesicles move primarily retrograde -- towards cis face.
b. Vesicles carry enzymes to modify and sort proteins. They do not carry the newly made proteins from the ER.
c. Sacs of Golgi move, carrying newly made proteins & lipids inside. New sacs are constantly formed at the cis face from material transported from the ER. Old sacs are lost from the trans face as they age.
d. Net Result: Enzyme composition of each sac changes with time. The enzyme composition of each individual sac is constantly changing as it ages and passes from cis to trans face. However the characteristic enzymes found in the sacs at each position of the Golgi (cis, medial & trans) remain the same, because the enzymes are "passed back." The vesicles retrieve the enzymes of "older" sacs (closer to the trans face) and carry them back to newer sacs (closer to the cis face).
3. Connection Model (FYI) -- another possibility is that the Golgi sacs are actually connected (although they seem to be separate), and that proteins move back and forth from one sac to the next, although net bulk flow of newly made proteins is from cis to trans. There isn't much evidence or enthusiasm for this model, but some recent data using genetically modified protein tagged with GFP has been interpreted in support of this model.
4. What really happens? The models above are not mutually exclusive, so a hybrid model is possible. Only new experiments generating new data will settle the question of how material actually moves. Not all materials may move through the Golgi in the same way. For a review of the models and the data as of 1998 (the 100th anniversary of the discovery of the Golgi) see Coming to Grips with the Golgi.
To review traffic through the Golgi, try problem 3-10. See also problems 3R-8 & 3R-9.
V. Lysosomes -- an example of sorting after the Golgi -- See Becker fig. 12-9 & Handout 13B
A. What's in a lysosome?
1. Lysosomes contain two classes of protein.
a. Enzymes -- Many different acid hydrolases that digest different macromolecules.
b. Substrates of the hydrolases -- Proteins to be degraded.
2. All proteins to be degraded in lysosomes are enclosed in a membrane.
a. Endo/Phagocytosis: Most proteins to be degraded in lysosomes come from outside the cell; enter vesicles by phagocytosis or endocytosis. See Sadava fig. 5.11 (4. 12).
b. Autophagy: Some proteins to be degraded come from the cytoplasm, and are encircled by internal membranes. (See 'autophagy' in texts if your are interested in more details. A picture is in Becker, fig. 12-22.)
3. How do two classes of protein come together? Vesicle or compartment containing substrate (proteins to be degraded) fuses with vesicle containing enzymes -- either a vesicle with newly made hydrolases or a pre-existing lysosome.
B. How do hydrolases get to lysosomes? Normal Pathway (See top of handout 13B. Steps refer to numbers on handout.)
1. How do hydrolases reach the Golgi?
2. How are hydrolases identified & tagged for transport to lysosomes?a. Enzyme synthesis: Hydrolases are made on ribosomes bound to ER. Enzymes enter ER co-translationally (as they are made).
b. Transport to Golgi: Hydrolases are transported in vesicles to Golgi. Steps 1 & 2.
a. Localization Signal -- Most hydrolases destined for lysosomes have a special sequence/patch.
b. Reading the LS -- Enzyme(s) recognize the sequence/patch and add Mannose-6-P (N-glycosylation starts in ER by addition of standard oligosaccharide. Modification of standard sugars to M6P occurs in Golgi -- this modification only happens to proteins with the proper amino acid sequence = soluble hydrolases bound for lysosomes) Step 3.
3. Role of M6P Receptor -- how is the tag recognized?
a. Binding the M6P -- Receptor in special part of trans Golgi binds proteins with M6P. Step 4.
b. Sorting in trans Golgi -- Proteins with M6P and their receptors accumulate in coated pits (step 5) and bud off (step 6) and go to a sorting vesicle/endosome (step 7).
c. Significance of this -- Many proteins are sorted in the Golgi, and the mechanism may be similar -- each class of cargo protein -- all those with a particular localization signal or tag -- may meet matching receptors in a particular part of the Golgi.
4. Sorting after the Golgi
a. Sorting vesicle/Endosome sorts multiple types of receptors and hydrolases. Same as what happens during RME when receptors and ligands are sorted. (step 7)
b. Recycling: M6P Receptors recycle back to Golgi (steps 8A & 10); vesicles with hydrolases add to old lysosome or form new one (steps 8B & 9). Note that 8A & 8B are equivalent to the same numbers on the handout of RME. 8B goes to the lysosome and 8A recycles back to the membrane from which it came.
c. SNAREs. (FYI) How do various vesicles fuse when they reach the proper target? There are matching transmembrane proteins (SNAREs) on the target membrane and the vesicle membrane. The cytoplasmic domains of the proteins are complementary, and pair up with each other. See SNARE hypothesis in Becker, p. 351-352 (348-349), if you are interested in more details.
C. Scavenger Pathway & Lysosomal diseases (See bottom of handout 13B)
1. I-cell disease (ICD) -- what happens if the enzyme that catalyzes formation of M6P is missing.
a. The primary defect: In I-cell disease, the defect is in the gene for an enzyme that modifies all the soluble hydrolases. So many hydrolases are affected, not just one. (Hydrolases that are membrane proteins are not affected.) Step 3 is skipped.
b. What happens to the hydrolases: All the soluble acid hydrolases lack M6P. So all the hydrolases that would normally stick to M6P receptors go to the wrong part of the Golgi. The hydrolases then end up in default vesicles. (as in Step 11) The default vesicles fuse with the plasma membrane and the hydrolases exit the cell. (Step 12) The hydrolases never reach the lysosomes.
c. The consequences: Inclusion bodies form = vesicles full of undigested materials that are normally degraded in lysosomes. ("Lysosomes" contain substrate, but no hydrolases to degrade the substrate.)
(1). Not all lysosomal enzymes are affected in I (inclusion) disease. This implies that there are other pathways for directing hydrolases to the lysosomes. (See problems 3-18 & 3R-1 for examples of alternative pathways.)(2). Not all tissues are affected in I cell disease. For example, the liver is not affected in ICD. This implies that in different tissues, either different pathways (to lysosomes) are used, and/or that different enzymes must be critical for proper lysosomal function.
2. Standard lysosomal storage diseases
a. What is a lysosomal storage disease? It's what happens if one hydrolase is missing or defective. A different enzyme is missing in each disease.
b. Examples: Gaucher's or Gaucher (pronounced 'Go-shay') disease & Tay-Sachs disease. In these cases, only one hydrolase is missing due to a defect in the gene for that enzyme. All other hydrolases reach the lysosomes and function normally.
c. How is I disease different? In I disease, most of the hydrolases are missing from lysosomes -- the hydrolases are made, but are not transported to lysosomes. The defect is not in the gene for a particular hydrolase. The defect is in a gene for a modification enzyme. This enzyme modifies the sugars attached to many different hydrolases.
d. Genetics: Each standard lysosomal storage disease or I disease is caused by a single, recessive mutation -- but the mutation in each disease is different.
3. Salvage (scavenger) pathway -- recovers normal hydrolases that accidentally end up outside the cell.
a. Some M6P receptors are "misdirected" : they are not recycled to Golgi -- instead they reach plasma membrane in some cells (by default pathway?). See dashed line in bottom of handout 13B.
b. Some normal hydrolases (with M6P) are "misdirected" -- they reach extra-cellular fluid (by default pathway?) -- "escape" from cell. (As in steps 11 and 12.)
c. Misplaced receptors can trap misplaced hydrolases: M6P receptors on the plasma membrane bind any extra-cellular hydrolases that "escaped" the cell (since these are normal hydrolases with M6P attached) . This is the "scavenger" part.
d. "Escaped" Hydrolases can be recovered by RME: Hydrolases bound to receptors are internalized by RME → endosome → lysosome = where they belonged in the first place. This is the "salvage" or recovery part.
e. Why the quotes? "Misdirected" and "escaped" are in quotes above, because this may be a normal pathway that some hydrolases always use to reach the lysosomes.
4. Use of Salvage Pathway to treat Gaucher's Disease -- Enzyme Replacement Therapy.
a. Missing lysosomal enzymes can be added: In cells with salvage pathway, added lysosomal hydrolases (containing M6P) can be taken up from outside. Hydrolases will be retrieved by RME and localized to lysosomes as explained above.
b. Practical Use: This method is currently used to treat Gaucher's disease (one hydrolase missing) at annual cost of $50,000 per patient for added enzyme. Enzyme is so expensive because M6P addition (& glycosylation in general) cannot yet be done in bacteria. Enzyme must be obtained from eukaryotic cells grown in tissue culture -- in bottles. Several other lysosomal diseases have been treated by enzyme replacement therapy at a similar cost per patient.
c. Can't treat I-cell disease this way. Note only one enzyme is being replaced here, not an entire set of hydrolases (as would be required to treat I-cell disease).
To review lysosomes and lysosomal diseases, try problem 3-9. See also 3-18 & 3R-1. For a catalog of all lysosomal diseases & current treatment see eMedicine. There are many additional articles in eMedicine about genetic & metabolic diseases.
VI. Lysosomes vs peroxisomes
- The two organelles are similar in structure but very different in enzyme content, function, and how they are put together. All details on peroxisomes in lecture 14.Next
Time
{kind=link}
{kind=link}