
Chapter 27
from:
web.virginia.edu/
The Synthesis and Degradation of Nucleotides

Pigeon drinking at Gaia Fountain, Siena, Italy. The basic features of purine
biosyn-
thesis were elucidated initially from metabolic studies of nitrogen metabolism
in
pigeons. Pigeons excrete excess N as uric acid, a purine analog. (Arte &
Imamagini
srl/Corbis Images.)
Nucleotides are ubiquitous constituents of life, actively participating in the majority of biochemical reactions. Recall that ATP is the “energy currency” of the cell, that uridine nucleotide derivatives of carbohydrates are common intermediates in cellular transformations of carbohydrates (Chapter 23), and that biosynthesis of phospholipids proceeds via cytosine nucleotide derivatives (Chapter 25). In Chapter 33, we will see that GTP serves as the immediate energy source driving the endergonic reactions of protein synthesis. Many of the coenzymes (such as coenzyme A, NAD, NADP, and FAD) are derivatives of nucleotides. Nucleotides also act in metabolic regulation, as in the response of key enzymes of intermediary metabolism to the relative concentrations of AMP, ADP, and ATP (PFK is a prime example here; see also Chapter 19). Further, cyclic derivatives of purine nucleotides, cAMP and cGMP, have no other role in metabolism than regulation. Last and not least, nucleotides are the monomeric units of nucleic acids. Deoxynucleoside triphosphates (dNTPs) and nucleoside triphosphates (NTPs) serve as the immediate substrates for the biosynthesis of DNA and RNA, respectively (see Part IV, Information Transfer). Without RNA, protein biosynthesis is not possible; in the absence of DNA synthesis, the genetic material is not replicated and cell division cannot occur.
27.1 · Nucleotide Biosynthesis
Nearly all organisms can make the purine and pyrimidine nucleotides via so-called de novo biosynthetic pathways. (De novo means “anew”; a less literal but more apt translation might be “from scratch” because de novo pathways are metabolic sequences that form complex end products from rather simple precursors.) Many organisms also have salvage pathways to recover purine and pyrimidine compounds obtained in the diet or released during nucleic acid turnover and degradation. While the ribose of nucleotides can be catabolized to generate energy, the nitrogenous bases do not serve as energy sources; their catabolism does not lead to products used by pathways of energy conservation. Compared to slowly dividing cells, rapidly proliferating cells synthesize larger amounts of DNA and RNA per unit time. To meet the increased demand for nucleic acid synthesis, substantially greater quantities of nucleotides must be produced. The pathways of nucleotide biosynthesis thus become attractive targets for the clinical control of rapidly dividing cells such as cancers or infectious bacteria. Many antibiotics and anticancer drugs are inhibitors of purine or pyrimidine nucleotide biosynthesis.27.2 ·
The
Biosynthesis of Purines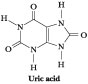
.Figure 27.1 · Nitrogen waste is excreted by birds principally as the purine analog, uric acid.
Substantial insight into
the de novo pathway for purine biosynthesis was provided in 1948 by John
Buchanan, who cleverly exploited the fact that birds excrete excess nitrogen
principally in the form of uric acid, a water-insoluble purine analog. Buchanan
fed isotopically labeled compounds to pigeons and then examined the distribution
of the labeled atoms in uric acid (Figure 27.1). By tracing the metabolic
source of the various atoms in this end product, he showed that the nine atoms
of the purine ring system (Figure 27.2) are contributed by aspartic acid (N-1),
glutamine (N-3 and N-9), glycine (C-4, C-5, and N-7), CO2 (C-6), and
THF one-carbon derivatives (C-2 and C-8). The coenzyme THF and its role in
one-carbon metabolism were introduced in
Chapter 18.
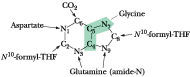
Figure 27.2 · The metabolic origin of the nine atoms in the purine ring system
 IMP
Biosynthesis: Inosinic Acid Is the Immediate Precursor to GMP and AMP
IMP
Biosynthesis: Inosinic Acid Is the Immediate Precursor to GMP and AMP
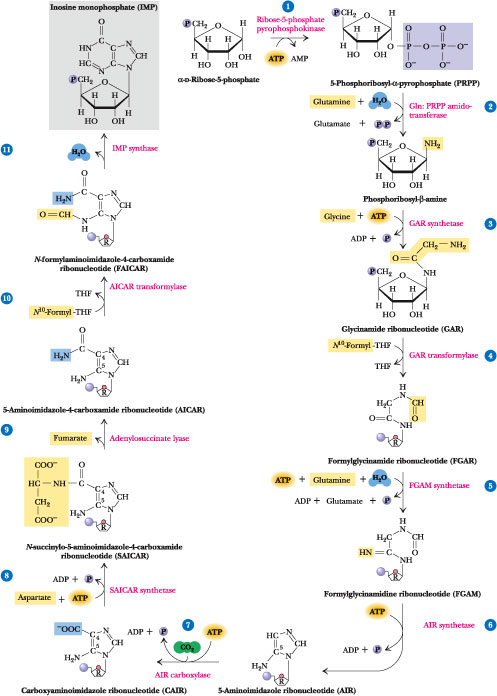
Figure 27.3 · The de novo pathway for purine synthesis. The first purine product of this pathway, IMP (inosinic acid or inosine monophosphate), serves as a precursor to AMP and GMP. Step 1: PRPP synthesis from ribose-5-phosphate and ATP by ribose-5-phosphate pyrophosphokinase. Step 2: 5-Phosphoribosyl-b-1-amine synthesis from a-PRPP, glutamine, and H2O by glutamine phosphoribosyl pyrophosphate amidotransferase. Step 3: Glycinamide ribonucleotide (GAR) synthesis from glycine, ATP, and 5-phosphoribosyl-b-amine by glycinamide ribonucleotide synthetase. Step 4: Formylglycinamide ribonucleotide synthesis from N10-formyl-THF and GAR by GAR transformylase. Step 5: Formylglycinamidine ribonucleotide (FGAM) synthesis from FGAR, ATP, glutamine, and H2O by FGAM synthetase (FGAR amidotransferase). The other products are ADP, Pi, and glutamate. Step 6: 5-Aminoimidazole ribonucleotide (AIR) synthesis is achieved via the ATP-dependent closure of the imidazole ring, as catalyzed by FGAM cyclase (AIR synthetase). (Note that the ring closure changes the numbering system.) Step 7: Carboxyaminoimidazole ribonucleotide (CAIR) synthesis from CO2, ATP, and AIR by AIR carboxylase. Step 8: N-succinylo-5-aminoimidazole-4-carboxamide ribonucleotide (SAICAR) synthesis from aspartate, CAIR, and ATP by SAICAR synthetase. Step 9: 5-Aminoimidazole carboxamide ribonucleotide (AICAR) formation by the nonhydrolytic removal of fumarate from SAICAR. The enzyme is adenylosuccinase. Step 10: 5-Formylaminoimidazole carboxamide ribonucleotide (FAICAR) formation from AICAR and N10-formyl-THF by AICAR transformylase. Step 11: Dehydration/ring closure yields the authentic purine ribonucleotide IMP. The enzyme is IMP synthase.
The de novo
synthesis of purines occurs in an interesting manner: The atoms forming the
purine ring are successively added to ribose-5-phosphate; thus, purines
are directly synthesized as nucleotide derivatives by assembling the atoms that
comprise the purine ring system directly on the ribose. In Step 1,
ribose-5-phosphate is activated via the direct transfer of a pyrophosphoryl
group from ATP to C-1 of the ribose, yielding
5-phosphoribosyl-a-pyrophosphate (PRPP) (Figure 27.3). The enzyme is
ribose-5-phosphate pyrophosphokinase. PRPP is the limiting substance in
purine biosynthesis. The two major purine nucleoside diphosphates, ADP and GDP,
are negative effectors of ribose-5-phosphate pyrophosphokinase. However, because
PRPP serves additional metabolic needs, the next reaction is actually the
committed step in the pathway.
Step 2 (Figure 27.3) is catalyzed by glutamine
phosphoribosyl pyrophosphate amidotransferase. The anomeric carbon of the
substrate PRPP is in the a-configuration; the product is a b-glycoside (recall
that all the biologically important nucleotides are b-glycosides). The N atom of
this N-glycoside becomes N-9 of the nine-membered purine ring; it is the
first atom added in the construction of this ring. Glutamine phosphoribosyl
pyrophosphate amidotransferase is subject to feedback inhibition by GMP, GDP,
 Figure
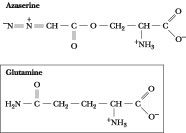
27.4 · The structure of
azaserine. Azaserine acts as an irreversible inhibitor of glutamine-dependent
enzymes by covalently attaching to nucleophilic groups in the glutamine-binding
site.
Figure
27.4 · The structure of
azaserine. Azaserine acts as an irreversible inhibitor of glutamine-dependent
enzymes by covalently attaching to nucleophilic groups in the glutamine-binding
site.
and GTP as well as AMP,
ADP, and ATP. The G series of nucleotides interacts at a guanine-specific
allosteric site on the enzyme, whereas the adenine nucleotides act at an
A-specific site. The pattern of inhibition by these nucleotides is competitive,
thus ensuring that residual enzyme activity is expressed until sufficient
amounts of both adenine and guanine nucleotides are synthesized. Glutamine
phosphoribosyl pyrophosphate amidotransferase is also sensitive to inhibition by
the glutamine analog azaserine (Figure 27.4). Azaserine has been employed
as an antitumor agent because it causes inactivation of glutamine-dependent
enzymes in the purine biosynthetic pathway.
Step 3 is carried out by glycinamide ribonucleotide
synthetase (GAR synthetase) via its ATP-dependent condensation of the
glycine carboxyl group with the amine of 5-phosphoribosyl-b amine (Figure
27.3). The reaction proceeds in two stages. First, the glycine carboxyl group is
activated via ATP-dependent phosphorylation. Next, an amide bond is formed
between the activated carboxyl of glycine and the b-amine. Glycine contributes
C-4, C-5, and N-7 of the purine.
Step 4 is the first of two THF-dependent reactions in the
purine pathway. GAR transformylase transfers the N10-formyl group of
N10-formyl-THF to the free amino group of GAR to yield
a-N-formylglycinamide ribonucleotide (FGAR). Thus, C-8 of the purine is
“formyl-ly” introduced. Although all of the atoms of the imidazole portion of
the purine ring are now present, the ring is not closed until Reaction 6.
Step 5 is catalyzed by FGAR amidotransferase (also
known as FGAM synthetase). ATP-dependent transfer of the glutamine amido
group to the C-4-carbonyl of FGAR yields formylglycinamidine ribonucleotide
(FGAM). As a glutamine-dependent enzyme, FGAR amidotransferase is, like
glutamine phosphoribosyl pyrophosphate amidotransferase (Reaction 2),
irreversibly inactivated by azaserine. The imino-N becomes N-3 of the purine.
Step 6 is an ATP-dependent dehydration that leads to
formation of the imidazole ring. ATP is used to phosphorylate the oxygen atom of
the formyl group, activating it for the ring closure step that follows. Because
the product is 5-aminoimidazole ribonucleotide, or AIR, this
enzyme is called AIR synthetase. In avian liver, the enzymatic activities
for Steps 3, 4, and 6 (GAR synthetase, GAR transformylase, and AIR synthetase)
reside on a single, 110-kD multifunctional polypeptide.
In Step 7, carbon dioxide is added at the C-4 position of
the imidazole ring by AIR carboxylase in an ATP-dependent reaction; the
carbon of CO2 will become C-6 of the purine ring. The product is
carboxyaminoimidazole ribonucleotide (CAIR).
In Step 8, the amino-N of aspartate provides N-1 through
linkage to the C-6 carboxyl function of CAIR. ATP hydrolysis drives the
condensation of Asp with CAIR. The product is
N-succinylo-5-aminoimidazole-4-carboxamide ribonucleotide (SAICAR).
SAICAR synthetase catalyzes the reaction. The enzymatic activities for Steps
7 and 8 reside on a single, bifunctional polypeptide in avian liver.
Step 9 removes the four carbons of Asp as fumaric acid in
a nonhydrolytic cleavage. The product is 5-aminoimidazole-4-carboxamide
ribonucleotide (AICAR); the enzyme is adenylosuccinase
(adenylosuccinate lyase). Adenylosuccinase acts again in that part of the
purine pathway leading from IMP to AMP and derives its name from this latter
activity (see following). AICAR is also an intermediate in the histidine
biosynthetic pathway (see
Chapter 26), but because the purine nucleotide ATP is the precursor to AICAR
in that pathway, no net purine synthesis is achieved.
Step 10 adds the formyl carbon of N10-formyl-THF
as the ninth and last atom necessary for forming the purine nucleus. The enzyme
is called AICAR transformylase; the products are THF and
N-formylaminoimidazole-4-carboxamide ribonucleotide, or FAICAR.
Step 11 involves dehydration and ring closure and
completes the initial phase of purine biosynthesis. The enzyme is IMP
cyclohydrolase (also known as IMP synthase and inoisinicase).
Unlike Step 6, this ring closure does not require ATP. In avian liver, the
enzymatic activities catalyzing Steps 10 and 11 (AICAR transformylase and
inoisinicase) activities reside on 67-kD bifunctional polypeptides organized
into 135-kD dimers.
 Figure
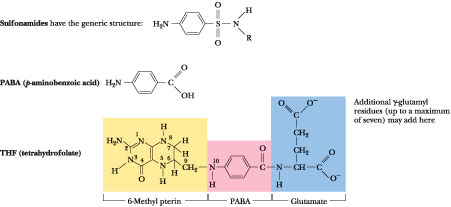
27.5 · Sulfa drugs, or
sulfonamides, owe their antibiotic properties to their similarity to p-aminobenzoate
(PABA), an important precursor in folic acid synthesis. Sulfonamides block folic
acid formation by competing with PABA.
Figure
27.5 · Sulfa drugs, or
sulfonamides, owe their antibiotic properties to their similarity to p-aminobenzoate
(PABA), an important precursor in folic acid synthesis. Sulfonamides block folic
acid formation by competing with PABA.
Note that six ATPs
are required in the purine biosynthetic pathway from ribose-5-phosphate to IMP:
one each at Steps 1, 3, 5, 6, 7, and 8. However, 7 high-energy phosphate bonds
(equal to 7 ATP equivalents) are consumed because a-PRPP formation in Reaction 1
followed by PPi release in Reaction 2 represents the loss of 2 ATP
equivalents.
The dependence of purine biosynthesis on folic acid
compounds at Steps 4 and 10 means that antagonists of folic acid metabolism (for
example, methotrexate; see Figure 27.30) indirectly
inhibit purine formation and, in turn, nucleic acid synthesis, cell growth, and
cell division. Clearly, rapidly dividing cells such as malignancies or infective
bacteria are more susceptible to these antagonists than slower-growing normal
cells. Also among the folic acid antagonists are sulfonamides (Figure
27.5). Folic acid is a vitamin for animals and is obtained in the diet. In
contrast, bacteria synthesize folic acid from precursors, including
p-aminobenzoic acid (PABA), and thus are more susceptible to sulfonamides
than are animal cells.
AMP and GMP Are Synthesized from IMP
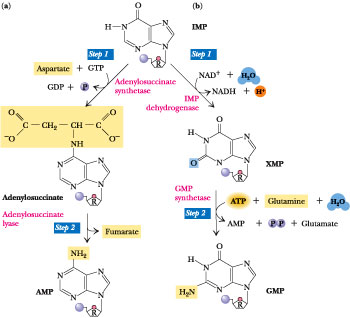 Figure
27.6 · The synthesis of AMP and
GMP from IMP. (a) AMP synthesis: The two reactions of AMP synthesis mimic Steps
8 and 9 in the purine pathway leading to IMP. In Step 1, the 6-O of
inosine is displaced by aspartate to yield adenylosuccinate. The energy required
to drive this reaction is derived from GTP hydrolysis. The enzyme is
adenylosuccinate synthetase. AMP is a competitive inhibitor (with respect to the
substrate IMP) of adenylosuccinate synthetase. In Step 2,
adenylosuccinase (also known as adenylosuccinate lyase, the same enzyme
catalyzing Step 9 in the purine pathway) carries out the nonhydrolytic removal
of fumarate from adenylosuccinate, leaving AMP. (b) GMP synthesis: The two
reactions of GMP synthesis are an NAD+-dependent oxidation followed
by an amidotransferase reaction. In Step 1, IMP dehydrogenase employs the
substrates NAD+ and H2O in catalyzing oxidation of IMP at
C-2. The products are xanthylic acid (XMP or xanthosine monophosphate), NADH,
and H+. GMP is a competitive inhibitor (with respect to IMP) of IMP
dehydrogenase. In Step 2, transfer of the amido-N of glutamine to the C-2
position of XMP yields GMP. This ATP-dependent reaction is catalyzed by GMP
synthetase. Besides GMP, the products are glutamate, AMP, and PPi.
Hydrolysis of PPi to two Pi by ubiquitous pyrophosphatases
pulls this reaction to completion.
Figure
27.6 · The synthesis of AMP and
GMP from IMP. (a) AMP synthesis: The two reactions of AMP synthesis mimic Steps
8 and 9 in the purine pathway leading to IMP. In Step 1, the 6-O of
inosine is displaced by aspartate to yield adenylosuccinate. The energy required
to drive this reaction is derived from GTP hydrolysis. The enzyme is
adenylosuccinate synthetase. AMP is a competitive inhibitor (with respect to the
substrate IMP) of adenylosuccinate synthetase. In Step 2,
adenylosuccinase (also known as adenylosuccinate lyase, the same enzyme
catalyzing Step 9 in the purine pathway) carries out the nonhydrolytic removal
of fumarate from adenylosuccinate, leaving AMP. (b) GMP synthesis: The two
reactions of GMP synthesis are an NAD+-dependent oxidation followed
by an amidotransferase reaction. In Step 1, IMP dehydrogenase employs the
substrates NAD+ and H2O in catalyzing oxidation of IMP at
C-2. The products are xanthylic acid (XMP or xanthosine monophosphate), NADH,
and H+. GMP is a competitive inhibitor (with respect to IMP) of IMP
dehydrogenase. In Step 2, transfer of the amido-N of glutamine to the C-2
position of XMP yields GMP. This ATP-dependent reaction is catalyzed by GMP
synthetase. Besides GMP, the products are glutamate, AMP, and PPi.
Hydrolysis of PPi to two Pi by ubiquitous pyrophosphatases
pulls this reaction to completion.
IMP is the precursor to
both AMP and GMP. These major purine nucleotides are formed via distinct
two-step metabolic pathways that diverge from IMP. The branch leading to AMP
(adenosine 5'-monophosphate) involves the displacement of the 6-O group of
inosine with aspartate (Figure 27.6) in a GTP-dependent reaction, followed by
the nonhydrolytic removal of the 4-carbon skeleton of Asp as fumarate; the Asp
amino group remains as the 6-amino group of AMP. Adenylosuccinate synthetase
and adenylosuccinase are the two enzymes. Recall that adenylosuccinase
also acted at Step 9 in the pathway from ribose-5-phosphate to IMP. Fumarate
production provides a connection between purine synthesis and the citric acid
cycle.
The formation of GMP from IMP requires oxidation at C-2 of
the purine ring, followed by a glutamine-dependent amidotransferase reaction
that replaces the oxygen on C-2 with an amino group to yield 2-amino,6-oxy
purine nucleoside monophosphate, or as this compound is commonly known,
guanosine monophosphate. The enzymes in the GMP branch are IMP
dehydrogenase and GMP synthetase. Note that, starting from
ribose-5-phosphate, 8 ATP equivalents are consumed in the synthesis of AMP and 9
in the synthesis of GMP.
Regulation of the Purine Biosynthetic Pathway
The regulatory network
that controls purine synthesis is schematically represented in Figure 27.7. To
recapitulate, the purine biosynthetic pathway from ribose-5-phosphate to IMP is
allosterically regulated at the first two steps. Ribose-5-phosphate
pyrophosphokinase, although not the committed step in purine synthesis, is
subject to feedback inhibition by ADP and GDP.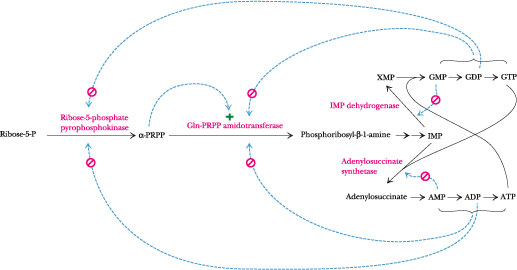
Figure 27.7 · The regulatory circuit controlling purine biosynthesis. ADP and GDP are feedback inhibitors of ribose-5-phosphate pyrophosphokinase, the first reaction in the pathway. The second enzyme, glutamine phosphoribosyl pyrophosphate amidotransferase, has two distinct feedback inhibition sites, one for A nucleotides and one for G nucleotides. Also, this enzyme is allosterically activated by PRPP. In the branch leading from IMP to AMP, the first enzyme is feedback-inhibited by AMP, while the corresponding enzyme in the branch from IMP to GMP is feedback-inhibited by GMP. Last, ATP is the energy source for GMP synthesis, whereas GTP is the energy source for AMP synthesis.
The enzyme catalyzing
the next step, glutamine phosphoribosyl pyrophosphate amidotransferase, has two
allosteric sites, one where the “A” series of nucleoside phosphates (AMP, ADP,
and ATP) binds and feedback-inhibits, and another where the corresponding “G”
series binds and inhibits. Further, PRPP is a “feed-forward” activator of this
enzyme. Thus, the rate of IMP formation by this pathway is governed by the
levels of the final end products, the adenine and guanine nucleotides.
The purine pathway splits at IMP. The first enzyme in the
AMP branch, adenylosuccinate synthetase, is competitively inhibited by AMP. Its
counterpart in the GMP branch, IMP dehydrogenase, is inhibited in like fashion
by GMP. Thus, the fate of IMP is determined by the relative levels of AMP and
GMP, so that any deficiency in the amount of either of the principal purine
nucleotides is self-correcting. This reciprocity of regulation is an effective
mechanism for balancing the formation of AMP and GMP to satisfy cellular needs.
Note also that reciprocity is even manifested at the level of energy input: GTP
provides the energy to drive AMP synthesis, while ATP serves this role in GMP
synthesis (Figure 27.7).
ATP-Dependent Kinases Form Nucleoside Diphosphates and Triphosphates from the Nucleoside Monophosphates
The products of de novo purine biosynthesis are the nucleoside monophosphates AMP and GMP. These nucleotides are converted by successive phosphorylation reactions into their metabolically prominent triphosphate forms, ATP and GTP. The first phosphorylation, to give the nucleoside diphosphate forms, is carried out by two base-specific, ATP-dependent kinases, adenylate kinase and guanylate kinase.
Adenylate kinase: AMP + ATP ® 2 ADP
Guanylate kinase: GMP + ATP ® GDP + ADP
These nucleoside monophosphate kinases also act on deoxynucleoside monophosphates to give dADP or dGDP.GDP + ATP GTP + ADP
Because this enzymatic reaction is readily reversible and nonspecific with respect to both phosphoryl acceptor and donor, in effect any NDP can be phosphorylated by any NTP, and vice versa. The preponderance of ATP over all other nucleoside triphosphates means that, in quantitative terms, it is the principal nucleoside diphosphate kinase substrate. The enzyme does not discriminate between the ribose moieties of nucleotides and thus functions in phosphoryl transfers involving deoxy-NDPs and deoxy-NTPs as well.
27.3 · Purine Salvage
Nucleic acid turnover
(synthesis and degradation) is an ongoing metabolic process in most cells.
Messenger RNA in particular is actively synthesized and degraded. These
degradative processes can lead to the release of free purines in the form of
adenine, guanine, and hypoxanthine (the base in IMP). These substances represent
a metabolic investment by cells. So-called salvage pathways exist to recover
them in useful form. Salvage reactions involve resynthesis of nucleotides from
bases via phosphoribosyltransferases.
Base + PRPP nucleoside-5'-phosphate + PPi
The subsequent
hydrolysis of PPi to inorganic phosphate by pyrophosphatases renders
the phosphoribosyltransferase reaction effectively irreversible.
The purine phosphoribosyltransferases are adenine
phosphoribosyltransferase (APRT), which mediates AMP formation, and
hypoxanthine-guanine phosphoribosyltransferase (HGPRT), which can act
on either hypoxanthine to form IMP or guanine to form GMP (shaded area, Figure
27.8).
Lesch-Nyhan Syndrome: HGPRT Deficiency Leads to Severe Clinical Disorder
The symptoms of Lesch-Nyhan syndrome are tragic: a crippling gouty arthritis due to excessive uric acid accumulation (uric acid is a purine degradation product, discussed in the next section) and, worse, severe malfunctions in the nervous system that lead to mental retardation, spasticity, aggressive behavior, and self-mutilation. Lesch-Nyhan syndrome results from a complete deficiency in HGPRT activity. The structural gene for HGPRT is located on the X chromosome, and the disease is a congenital, recessive, sex-linked trait manifested only in males. The severe consequences of HGPRT deficiency argue that purine salvage has greater metabolic importance than simply the energy-saving recovery of bases. Although HGPRT might seem to play a minor role in purine metabolism, its absence has profound consequences: de novo purine biosynthesis is dramatically increased and uric acid levels in the blood are elevated. Presumably, these changes ensue because lack of consumption of PRPP by HGPRT elevates its availability for glutamine-PRPP amidotransferase, enhancing overall de novo purine synthesis and, ultimately, uric acid production (Figure 27.8). Despite these explanations, it remains unclear why deficiency in this single enzyme leads to the particular neurological aberrations characteristic of the syndrome. Fortunately, deficiencies in HGPRT activity in fetal cells can be detected following amniocentesis.
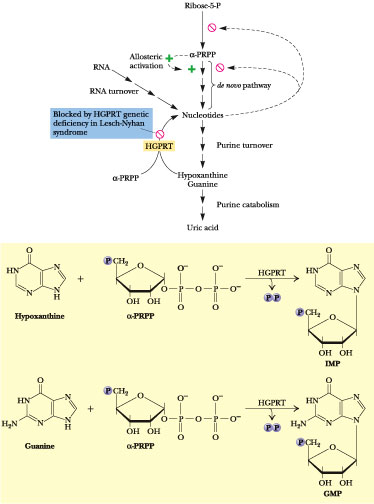
Figure 27.8 · The metabolic consequences of congenital HGPRT deficiency in Lesch-Nyhan syndrome. Loss of HGPRT leads to elevated PRPP levels and stimulation of de novo purine synthesis. One ultimate consequence is increased production of uric acid.
27.4 · Purine Degradation
Because nucleic acids are ubiquitous in cellular material, significant amounts are ingested in the diet. Nucleic acids are degraded in the digestive tract to nucleotides by various nucleases and phosphodiesterases. Nucleotides are then converted to
nucleosides by
base-specific nucleotidases and nonspecific phosphatases.
NMP + H2O ® nucleoside + Pi
Nucleosides are
hydrolyzed by nucleosidases or nucleoside phosphorylases to release the purine
base:
The pentoses liberated in these reactions provide the only
source of metabolic energy available from purine nucleotide degradation.
Feeding experiments using radioactively labeled nucleic
acids as metabolic tracers have demonstrated that little of the nucleotide
ingested in the diet is incorporated into cellular nucleic acids. These findings
confirm the de novo pathways of nucleotide biosynthesis as the primary
source of nucleic acid precursors. Ingested bases are, for the most part,
excreted. Nevertheless, cellular nucleic acids do undergo degradation in the
course of the continuous recycling of cellular constituents.
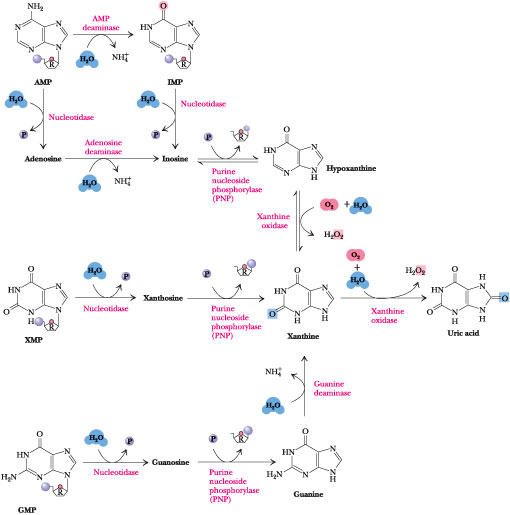
Figure 27.9 · The major pathways for purine catabolism in animals. Catabolism of the different purine nucleotides converges in the formation of uric acid.
The Major Pathways of Purine Catabolism Lead to Uric Acid
The major pathways of purine catabolism in animals are outlined in Figure 27.9. The various nucleotides are first converted to nucleosides by intracellular nucleotidases. These nucleotidases are under strict metabolic regulation so that their substrates, which act as intermediates in many vital processes, are not depleted below critical levels. Nucleosides are then degraded by the enzyme purine nucleoside phosphorylase (PNP) to release the purine base and ribose-l-P. Note that neither adenosine nor deoxyadenosine is a substrate for PNP. Instead, these nucleosides are first converted to inosine by adenosine deaminase. The PNP products are merged into xanthine by guanine deaminase and xanthine oxidase, and xanthine is then oxidized to uric acid by this latter enzyme.
Severe Combined Immunodeficiency Syndrome: A Lack of Adenosine Deaminase Is One Cause of This Inherited Disease
Severe combined
immunodeficiency syndrome, or SCID, is a group of related inherited
disorders characterized by the lack of an immune response to infectious disease.
This immunological insufficiency is attributable to the inability of B and T
lymphocytes to proliferate and produce antibodies in reaction to an antigenic
challenge. About 30% of SCID patients suffer from a deficiency in the enzyme
adenosine deaminase (ADA). ADA deficiency is also implicated in a variety of
other diseases, including AIDS, anemia, and various lymphomas and leukemias.
Gene therapy, the repair of a genetic deficiency by introduction of a
functional recombinant version of the gene, has been attempted on
individuals with SCID due to a defective ADA gene. ADA is a Zn2+-dependent
enzyme, and Zn2+ deficiency can also lead to reduced immune function.
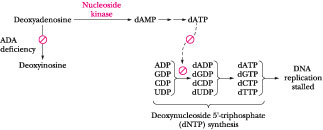 Figure
27.10 ·
The effect of elevated levels of deoxyadenosine on
purine metabolism. If ADA is deficient or absent, deoxyadenosine is not
converted into deoxyinosine as normal (see Figure 27.7). Instead, it is salvaged
by a nucleoside kinase, which converts it to dAMP, leading to accumulation of
dATP and inhibition of deoxynucleotide synthesis (see Figure
27.26). Thus, DNA replication is stalled.
Figure
27.10 ·
The effect of elevated levels of deoxyadenosine on
purine metabolism. If ADA is deficient or absent, deoxyadenosine is not
converted into deoxyinosine as normal (see Figure 27.7). Instead, it is salvaged
by a nucleoside kinase, which converts it to dAMP, leading to accumulation of
dATP and inhibition of deoxynucleotide synthesis (see Figure
27.26). Thus, DNA replication is stalled.
In the absence of ADA, deoxyadenosine is not degraded but
instead is converted into dAMP and then into dATP. dATP is a potent feedback
inhibitor of deoxynucleotide biosynthesis (discussed later in this chapter).
Without deoxyribonucleotides, DNA cannot be replicated and cells cannot divide (Figure
27.10). Rapidly proliferating cell types such as lymphocytes are
particularly susceptible if DNA synthesis is impaired.
The Purine Nucleoside Cycle: An Anaplerotic Pathway in Skeletal Muscle
Deamination of AMP to IMP by AMP deaminase (Figure 27.9) followed by resynthesis of AMP from IMP by the de novo purine pathway enzymes, adenylosuccinate synthetase and adenylosuccinate lyase, constitutes a purine nucleoside cycle (Figure 27.11). This cycle has the net effect of converting aspartate to fumarate plus NH4+. Although this cycle might seem like senseless energy consumption, it plays an important role in energy metabolism in skeletal muscle: the fumarate that it generates replenishes the levels of citric acid cycle intermediates lost in amphibolic side reactions (see Chapter 20). Skeletal muscle lacks the usual complement of anaplerotic enzymes and relies on enhanced levels of AMP deaminase, adenylosuccinate synthetase, and adenylosuccinate lyase to compensate.
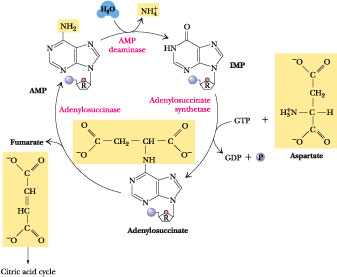
Figure 27.11 ·
The purine nucleoside cycle for anaplerotic replenishment of citric acid
cycle intermediates in skeletal muscle.
Xanthine Oxidase
Xanthine oxidase
(Figure 27.9) is present in large amounts in liver, intestinal
mucosa, and milk. It oxidizes hypoxanthine to xanthine and xanthine to uric
acid. Xanthine oxidase is a rather indiscriminate enzyme, using molecular oxygen
to oxidize a wide variety of purines, pteridines, and aldehydes, producing H2O2
as a product. Xanthine oxidase possesses FAD, nonheme Fe-S centers, and a
molybdenum cofactor (see
Figure 26.2)
as electron-transferring prosthetic groups. Its mechanism of action is
diagrammed in Figure 27.12).
In humans and other primates, uric acid is the end product
of purine catabolism and is excreted in the urine. Birds, terrestrial reptiles,
and many insects also excrete uric acid, but, in these organisms, uric acid
represents the major nitrogen excretory compound, because, unlike mammals, they
do not also produce urea (Chapter
26). Instead, the catabolism of all nitrogenous compounds, including amino
acids, is channeled into uric acid. This route of nitrogen catabolism allows
these animals to conserve water by excreting crystals of uric acid in paste-like
solid form.
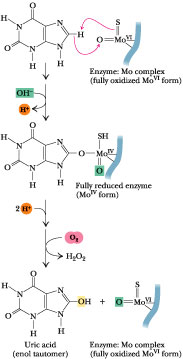
Figure 27.12 · Xanthine oxidase catalyzes a hydroxylase-type reaction.
Gout: An Excess of Uric Acid
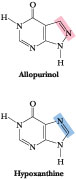
Figure 27.13 · Allopurinol, an analog of hypoxanthine, is a potent inhibitor of xanthine oxidase.
Gout is the clinical term describing the physiological consequences accompanying excessive uric acid accumulation in body fluids. Uric acid and urate salts are rather insoluble in water and tend to precipitate from solution if produced in excess. The most common symptom of gout is arthritic pain in the joints as a result of urate deposition in cartilaginous tissue. The joint of the big toe is particularly susceptible. Urate crystals may also appear as kidney stones and lead to painful obstruction of the urinary tract. Hyperuricemia, chronic elevation of blood uric acid levels, occurs in about 3% of the population as a consequence of impaired excretion of uric acid or overproduction of purines. Purine-rich foods (such as caviar—fish eggs rich in nucleic acids) may exacerbate the condition. The biochemical causes of gout are varied. However, a common treatment is allopurinol (Figure 27.13). This hypoxanthine analog binds tightly to xanthine oxidase, thereby inhibiting its activity and preventing uric acid formation. Hypoxanthine and xanthine do not accumulate to harmful concentrations because they are more soluble and thus more easily excreted.
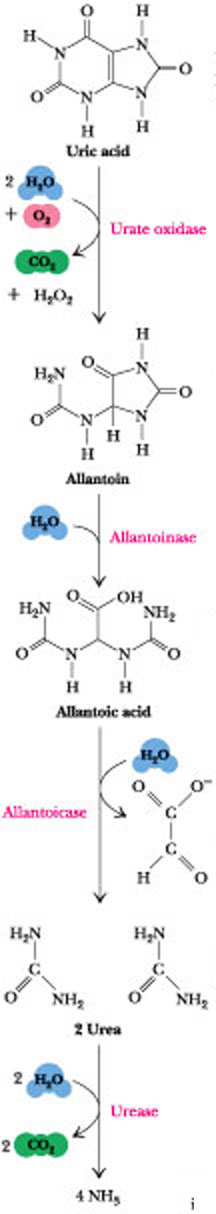
Figure 27.14 · The catabolism of uric acid to allantoin, allantoic acid, urea, or ammonia in various animals.
The Fate of Uric Acid
The subsequent metabolism of uric acid in organisms that don’t excrete it is shown in Figure 27.14. In molluscs and in mammals other than primates, uric acid is oxidized by urate oxidase to allantoin and excreted. In bony fishes (teleosts), uric acid degradation proceeds through yet another step wherein allantoin is hydrolyzed to allantoic acid by allantoinase before excretion. Cartilaginous fish (sharks and rays) as well as amphibians further degrade allantoic acid via the enzyme, allantoicase, to liberate glyoxylic acid and two equivalents of urea. Even simpler animals, such as most marine invertebrates (crustacea and so forth), use urease to hydrolyze urea to CO2 and ammonia. In contrast to animals that must rid themselves of potentially harmful nitrogen waste products, microorganisms often are limited in growth by nitrogen availability. Many possess an identical pathway of uric acid degradation, using it instead to liberate NH3 from uric acid so that it can be assimilated into organic-N compounds essential to their survival.
27.5 · The Biosynthesis of Pyrimidines
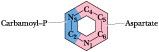 Figure
27.15 ·
The metabolic origin of the six atoms of the pyrimidine ring.
Figure
27.15 ·
The metabolic origin of the six atoms of the pyrimidine ring.
In contrast to purines,
pyrimidines are not synthesized as nucleotide derivatives. Instead, the
pyrimidine ring system is completed before a ribose-5-P moiety is attached.
Also, only two precursors, carbamoyl-P and aspartic acid, contribute atoms to
the six-membered pyrimidine ring (Figure 27.15), compared to
seven precursors for the 9 purine atoms.
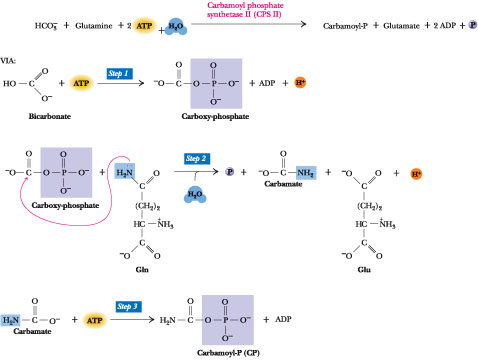 Figure
27.16 ·
The reaction catalyzed by carbamoyl phosphate synthetase II (CPS II). Note
that, in contrast to carbamoyl phosphate synthetase I, CPS II uses the amide of
glutamine, not NH4+, to form carbamoyl-P. Step 1:
The first ATP consumed in carbamoyl phosphate synthesis is used in forming
carboxy-phosphate, an activated form of CO2. Step 2:
Carboxy-phosphate (also called carbonyl-phosphate) then reacts with the
glutamine amide to yield carbamate and glutamate. Step 3: Carbamate is
phosphorylated by the second ATP to give ADP and carbamoyl phosphate.
Figure
27.16 ·
The reaction catalyzed by carbamoyl phosphate synthetase II (CPS II). Note
that, in contrast to carbamoyl phosphate synthetase I, CPS II uses the amide of
glutamine, not NH4+, to form carbamoyl-P. Step 1:
The first ATP consumed in carbamoyl phosphate synthesis is used in forming
carboxy-phosphate, an activated form of CO2. Step 2:
Carboxy-phosphate (also called carbonyl-phosphate) then reacts with the
glutamine amide to yield carbamate and glutamate. Step 3: Carbamate is
phosphorylated by the second ATP to give ADP and carbamoyl phosphate.
Mammals have two
enzymes for carbamoyl phosphate synthesis. Carbamoyl phosphate for pyrimidine
biosynthesis is formed by carbamoyl phosphate synthetase II (CPS II), a
cytosolic enzyme. Recall that carbamoyl phosphate synthetase I is a
mitochondrial enzyme dedicated to the urea cycle and arginine biosynthesis (Chapter
26). The substrates of carbamoyl phosphate synthetase II are HCO3-,
H2O, glutamine, and 2 ATPs (Figure 27.16). Because
carbamoyl phosphate made by CPS II in mammals has no fate other than
incorporation into pyrimidines, mammalian CPS II can be viewed as the committed
step in the pyrimidine de novo pathway. Bacteria have but one CPS, and
its carbamoyl phosphate product is incorporated into arginine as well as
pyrimidines. Thus, the committed step in bacterial pyrimidine synthesis is the
next reaction, which is mediated by aspartate transcarbamoylase (ATCase).
ATCase catalyzes the condensation of carbamoyl phosphate
with aspartate to form carbamoyl-aspartate (Figure 27.17). No
ATP input is required at this step because carbamoyl phosphate represents an
“activated” carbamoyl group.
Step 3 of pyrimidine synthesis involves ring closure and
dehydration via linkage of the -NH2 group introduced by carbamoyl-P
with the former b-COO- of aspartate; this reaction is mediated by the
enzyme dihydroorotase. The product of the reaction is dihydroorotate, a
six-membered ring compound. Dihydroorotate is not a true pyrimidine, but its
oxidation yields orotate, which is. This oxidation (Step 4) is catalyzed
by dihydroorotate dehydrogenase. Bacterial dihydroorotate dehydrogenases
are NAD+-linked flavoproteins, which are somewhat unusual in
possessing both FAD and FMN; these enzymes also have nonheme Fe-S centers as
additional redox prosthetic groups. The eukaryotic version of dihydroorotate
dehydrogenase is a protein component of the inner mitochondrial membrane; its
immediate e- acceptor is a quinone, and the reducing
equivalents drawn from dihydroorotate can be used to drive ATP synthesis via
oxidative phosphorylation. At this stage, ribose-5-phosphate is joined to N-1 of
orotate, giving the pyrimidine nucleotide orotidine-5'-monophosphate, or
OMP (Step 5, Figure 27.17). The ribose phosphate donor
is PRPP; the enzyme is orotate phosphoribosyltransferase. The next
reaction is catalyzed by OMP decarboxylase. Decarboxylation of OMP gives
UMP (uridine-5'-monophosphate, or uridylic acid), one of the two
common pyrimidine ribonucleotides.
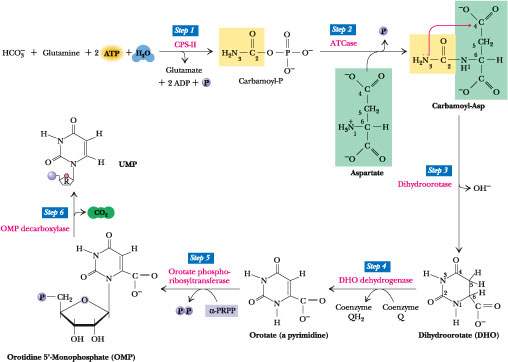
Figure 27.17 · The de novo pyrimidine biosynthetic pathway. Step 1: Carbamoyl-P synthesis. Step 2: Condensation of carbamoyl phosphate and aspartate to yield carbamoyl-aspartate is catalyzed by aspartate transcarbamoylase (ATCase). Step 3: An intramolecular condensation catalyzed by dihydroorotase gives the six-membered heterocyclic ring characteristic of pyrimidines. The product is dihydroorotate (DHO). Step 4: The oxidation of DHO by dihydroorotate dehydrogenase gives orotate. (In bacteria, NAD+ is the electron acceptor from DHO.) Step 5: PRPP provides the ribose-5-P moiety that transforms orotate into orotidine-5'-monophosphate, a pyrimidine nucleotide. Note that orotate phosphoribosyltransferase joins N-1 of the pyrimidine to the ribosyl group in appropriate b-configuration. PPi hydrolysis renders this reaction thermodynamically favorable. Step 6: Decarboxylation of OMP by OMP decarboxylase yields UMP.
Pyrimidine Biosynthesis in Mammals Is Another Example of “Metabolic Channeling”
In bacteria, the six
enzymes of de novo pyrimidine biosynthesis exist as distinct proteins,
each independently catalyzing its specific step in the overall pathway. In
contrast, in mammals, the six enzymatic activities are distributed among only
three proteins, two of which are multifunctional polypeptides: single
polypeptide chains having two or more enzymic centers. The first three steps of
pyrimidine synthesis, CPS-II, aspartate transcarbamoylase, and dihydroorotase,
are all localized on a single 210-kD cytosolic polypeptide. This multifunctional
enzyme is the product of a solitary gene, yet it is equipped with the active
sites for all three enzymatic activities. Step 4 (Figure 27.17)
is catalyzed by DHO dehydrogenase, a separate enzyme associated with the outer
surface of the inner mitochondrial membrane, but the enzymatic activities
mediating Steps 5 and 6, namely, orotate phosphoribosyltransferase and OMP
decarboxylase in mammals, are also found on a single cytosolic polypeptide known
as UMP synthase.
The purine biosynthetic pathway of avian liver also
provides examples of metabolic channeling. Recall that Steps 3, 4, and 6 of
de novo purine synthesis are catalyzed by three enzymatic activities
localized on a single multifunctional polypeptide, and Steps 7 and 8 and Steps
10 and 11 by respective bifunctional polypeptides (see Figure 27.3).
Such multifunctional enzymes confer an advantage: The product of one reaction in a pathway is the substrate for the next. In multifunctional enzymes, such products remain bound and are channeled directly to the next active site, rather than dissociated into the surrounding medium for diffusion to the next enzyme. This metabolic channeling is more efficient because substrates are not diluted into the milieu and no pools of intermediates accumulate. Synthesis of the Prominent Ribonucleotides UTP and CTP
The two prominent pyrimidine ribonucleotide products are derived from UMP via the same unbranched pathway. First, UDP is formed from UMP via an ATP-dependent nucleoside monophosphate kinase.
UMP + ATP 34UDP + ADP
Then, UTP is formed by nucleoside diphosphate kinase.
UDP + ATP 34UTP + ADP
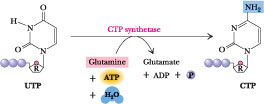 Figure
27.18 ·
CTP synthesis from UTP. CTP synthetase catalyzes amination of the
4-position of the UTP pyrimidine ring, yielding CTP. In eukaryotes, this NH2
comes from the amide-N of glutamine; in bacteria, NH4+
serves this role.
Figure
27.18 ·
CTP synthesis from UTP. CTP synthetase catalyzes amination of the
4-position of the UTP pyrimidine ring, yielding CTP. In eukaryotes, this NH2
comes from the amide-N of glutamine; in bacteria, NH4+
serves this role.
Amination of UTP at the 6-position gives CTP. The enzyme, CTP synthetase, is a glutamine amidotransferase (Figure 27.18). ATP hydrolysis provides the energy to drive the reaction.
Regulation of Pyrimidine Biosynthesis
Pyrimidine biosynthesis
in bacteria is allosterically regulated at aspartate trans-carbamoylase
(ATCase). Escherichia coli ATCase is feedback-inhibited by the end
product, CTP. ATP, which can be viewed as a signal of both energy availability
and purine sufficiency, is an allosteric activator of ATCase. CTP and ATP
compete for a common allosteric site on the enzyme. In many bacteria, UTP, not
CTP, acts as the ATCase feedback inhibitor.
In animals, CPS-II catalyzes the committed step in
pyrimidine synthesis and serves as the focal point for allosteric regulation.
UDP and UTP are feedback inhibitors of CPS-II, while PRPP and ATP are allosteric
activators. With the exception of ATP, none of these compounds are substrates of
CPS-II or of either of the two other enzymic activities residing with it on the
trifunctional polypeptide. Figure 27.19 compares the regulatory circuits
governing pyrimidine synthesis in bacteria and animals.
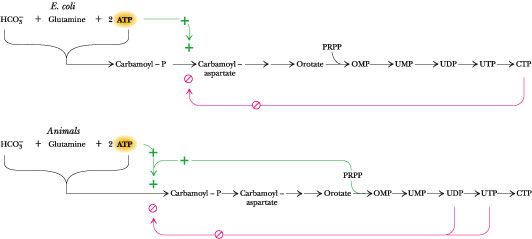 Figure 27.19 ·
A comparison of the regulatory circuits that control
pyrimidine synthesis in E. coli and animals.
Figure 27.19 ·
A comparison of the regulatory circuits that control
pyrimidine synthesis in E. coli and animals.
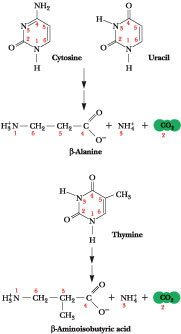
Figure 27.20 · Pyrimidine degradation. Carbons 4, 5, and 6 plus N-1 are released as b-alanine, N-3 as NH4+, and C-2 as CO2. (The pyrimidine thymine yields b-aminoisobutyric acid.) Recall that aspartate was the source of N-1 and C-4, -5, and -6, while C-2 came from CO2 and N-3 from NH4+ via glutamine.
27.6 · Pyrimidine Degradation
Like purines, free
pyrimidines can be salvaged and recycled to form nucleotides via
phosphoribosyltransferase reactions similar to those discussed earlier.
Pyrimidine catabolism results in degradation of the pyrimidine ring to products
reminiscent of the original substrates, aspartate, CO2, and ammonia
(Figure 27.20). b-Alanine can be recycled into the synthesis of coenzyme A.
Catabolism of the pyrimidine base, thymine (5-methyluracil) yields b-amino-isobutyric
acid instead of b-alanine.
Pathways presented thus far in this chapter account for
the synthesis of the four principal ribonucleotides: ATP, GTP, UTP, and CTP.
These compounds serve important coenzymic functions in metabolism and are the
immediate precursors for ribonucleic acid (RNA) synthesis. Roughly 90% of the
total nucleic acid in cells is RNA, with the remainder being deoxyribonucleic
acid (DNA). DNA differs from RNA in being a polymer of deoxyribonucleotides, one
of which is deoxythymidylic acid. We now turn to the synthesis of these
compounds.
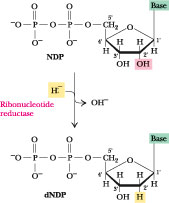 Figure
27.21 ·
Deoxyribonucleotide synthesis involves reduction at the 2'-position of the
ribose ring of nucleoside diphosphates.
Figure
27.21 ·
Deoxyribonucleotide synthesis involves reduction at the 2'-position of the
ribose ring of nucleoside diphosphates.
27.7 · Deoxyribonucleotide Biosynthesis
The deoxyribonucleotides
have only one metabolic purpose: to serve as precursors for DNA synthesis. In
most organisms, ribonucleoside diphosphates (NDPs) are the substrates for
deoxyribonucleotide formation. Reduction at the 2'-position of the ribose ring
in NDPs produces 2'-deoxy forms of these nucleotides (Figure
27.21). This reaction involves replacement of the 2'-OH by a hydride ion (H:-)
and is catalyzed by an enzyme known as ribonucleotide reductase.
Enzymatic ribonucleotide reduction involves a free radical mechanism, and three
classes of ribonucleotide reductases are known, differing from each other in
their mechanisms of free radical generation. Class I enzymes, found in E.
coli and virtually all eukaryotes, are Fe-dependent and generate the
required free radical on a specific tyrosyl side chain.
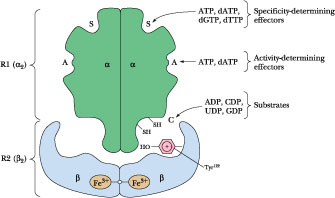 Figure
27.22 ·
E. coli ribonucleotide reductase: its binding sites and subunit
organization. Two proteins, R1 and R2 (each a dimer of identical subunits),
combine to form the holoenzyme. The holoenzyme has three classes of nucleotide
binding sites: S, the specificity-determining sites; A, the activity-determining
sites; and C, the catalytic or active site. These various sites bind different
nucleotide ligands. Note that the holoenzyme apparently possesses only one
active site formed by interaction between Fe3+ atoms in each R2
subunit.
Figure
27.22 ·
E. coli ribonucleotide reductase: its binding sites and subunit
organization. Two proteins, R1 and R2 (each a dimer of identical subunits),
combine to form the holoenzyme. The holoenzyme has three classes of nucleotide
binding sites: S, the specificity-determining sites; A, the activity-determining
sites; and C, the catalytic or active site. These various sites bind different
nucleotide ligands. Note that the holoenzyme apparently possesses only one
active site formed by interaction between Fe3+ atoms in each R2
subunit.
E. coli Ribonucleotide Reductase
The enzyme system for dNDP formation consists of four proteins, two of which constitute the ribonucleotide reductase proper, an enzyme of the a2b2 type. The other two proteins, thioredoxin and thioredoxin reductase, function in the delivery of reducing equivalents, as we shall see shortly. The two proteins of ribonucleotide reductase are designated R1 (86 kD) and R2 (43.5 kD) and each is a homodimer in the holoenzyme (Figure 27.22). The R1 homodimer carries two types of regulatory sites in addition to the catalytic site. Substrates (ADP, CDP, GDP, UDP) bind at the catalytic site. One regulatory site—the substrate specificity site—binds ATP, dATP, dGTP, or dTTP, and which of these nucleotides is bound there determines which nucleoside diphosphate is bound at the catalytic site. The other regulatory site, the overall activity site, binds either the activator ATP or the negative effector dATP; the nucleotide bound here determines whether the enzyme is active or inactive. Activity depends also on residues Cys439, Cys225, and Cys462 in R1. The 2 Fe atoms within the single active site formed by the R2 homodimer generate the free radical required for ribonucleotide reduction on a specific R2 residue, Tyr122, which in turn generates a thiyl free radical (Cys-S×) on Cys439. Cys439-S× initiates ribonucleotide reduction by abstracting the 3'-H from the ribose ring of the nucleoside diphosphate substrate (Figure 27.23) and forming a free radical on C-3'. Subsequent dehydration forms the deoxyribonucleotide product.
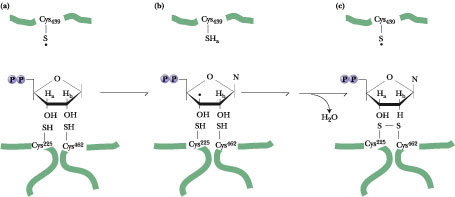
Figure 27.23 · The free radical mechanism of ribonucleotide reduction. Ha designates the C-3' hydrogen and Hb the C-2' hydrogen atom. Formation of a thiyl radical on Cys439 (a) of the E. coli ribonucleotide reductase R1 homodimer through reaction with a Tyr122 free radical on R2 leads to removal of the Ha hydrogen and creation of a C-3'× radical (b). Dehydration via removal of Hb together with the C-2'-OH group and restoration of Ha to C-3' forms the dNDP product, accompanied by oxidation of R1 Cys225 and Cys462OSH groups to form a disulfide (c). (Adapted from Reichard, P., 1997. The evolution of ribonucleotide reduction. Trends in Biochemical Sciences 22:81-85. This free radical mechanism of ribonucleotide reduction was originally proposed by JoAnn Stubbe of MIT.)
The Reducing Power for Ribonucleotide Reductase
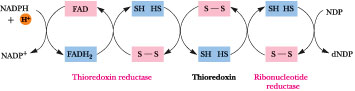
NADPH is the ultimate source of reducing equivalents for ribonucleotide reduction, but the immediate source is reduced thioredoxin, a small (12 kD) protein with reactive Cys-sulfhydryl groups situated next to one another in the sequence Cys-Gly-Pro-Cys. These Cys residues are able to undergo reversible oxidation-reduction between (-S-S-) and (-SH HS-) and, in their reduced form, serve as primary electron donors to regenerate the reactive -SH pair of the ribonucleotide reductase active site (Figure 27.23). In turn, the sulfhydryls of thioredoxin must be restored to the (-SH HS-) state for another catalytic cycle. Thioredoxin reductase, an a2-type enzyme composed of 58-kD flavoprotein subunits, mediates the NADPH-dependent reduction of thioredoxin (Figure 27.24). Thioredoxin functions in a number of metabolic roles besides deoxyribonucleotide synthesis, the common denominator of which is reversible sulfide:sulfhydryl transitions. Another sulfhydryl protein similar to thioredoxin, called glutaredoxin, can also function in ribonucleotide reduction. Oxidized glutaredoxin is re-reduced by two equivalents of glutathione (g-glutamylcysteinylglycine; Figure 27.25), which in turn is re-reduced by glutathione reductase, another NADPH-dependent flavoenzyme.
Figure 27.24 · he (-S-S-)/(-SH HS-) oxidation-reduction cycle involving ribonucleotide reductase, thioredoxin, thio-redoxin reductase, and NADPH.
The substrates for ribonucleotide reductase are CDP, UDP,
GDP, and ADP, and the corresponding products are dCDP, dUDP, dGDP, and dADP.
Because CDP is not an intermediate in pyrimidine nucleotide synthesis, it must
arise by dephosphorylation of CTP, for instance, via nucleoside diphosphate
kinase action. Although uridine nucleotides do not occur in DNA, UDP is a
substrate. The formation of dUDP is justified because it is a precursor to dTTP,
a necessary substrate for DNA synthesis (see following discussion).
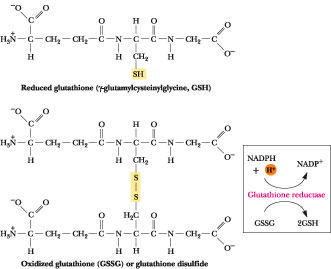 Figure 27.25 ·
The structure of glutathione.
Figure 27.25 ·
The structure of glutathione.
Regulation of Ribonucleotide Reductase Specificity and Activity
Ribonucleotide reductase activity must be modulated in two ways in order to maintain an appropriate balance of the four deoxynucleotides essential to DNA synthesis, namely, dATP, dGTP, dCTP, and dTTP. First, the overall activity of the enzyme must be turned on and off in response to the need for dNTPs. Second, the relative amounts of each NDP substrate transformed into dNDP must be controlled in order that the right balance of dATP:dGTP:dCTP:dTTP is produced. The two different sets of effector binding sites on ribonucleotide reductase, discrete from the substrate-binding active site, are designed to serve these purposes. These two regulatory sites are designated the overall activity site and the substrate specificity site. Only ATP and dATP are able to bind at the overall activity site. If ATP is bound, the enzyme is active, while if its deoxy counterpart, dATP, occupies this site, the enzyme is inactive. That is, ATP is a positive effector and dATP is a negative effector with respect to enzyme activity, and they compete for the same site.
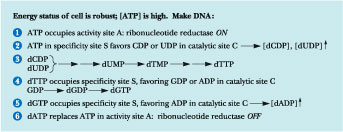 Figure
27.26 ·
Regulation of deoxynucleotide biosynthesis: The rationale for the various
affinities displayed by the two nucleotide-binding regulatory sites on
ribonucleotide reductase.
Figure
27.26 ·
Regulation of deoxynucleotide biosynthesis: The rationale for the various
affinities displayed by the two nucleotide-binding regulatory sites on
ribonucleotide reductase.
The second effector site, the substrate specificity site, can bind either ATP, dTTP, dGTP, or dATP, and the substrate specificity of the enzyme is determined by which of these nucleotides occupies this site. If ATP is in the substrate specificity site, ribonucleotide reductase preferentially binds pyrimidine nucleotides (UDP or CDP) at its active site and reduces them to dUDP and dCDP. With dTTP in the specificity-determining site, GDP is the preferred substrate. When dGTP binds to this specificity site, ADP becomes the favored substrate for reduction. The rationale for these varying affinities is as follows (Figure 27.26): High [ATP] is consistent with cell growth and division and, consequently, the need for DNA synthesis. Thus, ATP binds in the activity-determining site of ribonucleotide reductase, turning it on and promoting production of dNTPs for DNA synthesis. Under these conditions, ATP is also likely to occupy the substrate specificity site, so that UDP and CDP are reduced to dUDP and dCDP. As we shall soon see, both of these pyrimidine deoxynucleoside diphosphates are precursors to dTTP. Thus, elevation of dUDP and dCDP levels leads to an increase in [dTTP]. High dTTP levels increase the likelihood that it will occupy the substrate specificity site, in which case GDP becomes the preferred substrate, and dGTP levels rise. Upon dGTP association with the substrate specificity site, ADP is the favored substrate, leading to ADP reduction and the eventual accumulation of dATP. Binding of dATP to the overall activity site then shuts the enzyme down. In summary, the relative affinities of the three classes of nucleotide binding sites in ribonucleotide reductase for the various substrates, activators, and inhibitors are such that the formation of dNDPs proceeds in an orderly and balanced fashion. As these dNDPs are formed in amounts consistent with cellular needs, their phosphorylation by nucleoside diphosphate kinases produces dNTPs, the actual substrates of DNA synthesis.
27.8 · Synthesis of Thymine Nucleotides
![]() Figure
27.27 ·
Pathways of dTMP synthesis. dTMP production is dependent on dUMP formation
from dCDP and dUDP synthesis. If the dCDP pathway is traced from the common
pyrimidine precursor, UMP, it will proceed as follows: UMP ®
UDP ® UTP ® CTP
® CDP ® dCDP
® dCMP ® dUMP
® dTMP.
Figure
27.27 ·
Pathways of dTMP synthesis. dTMP production is dependent on dUMP formation
from dCDP and dUDP synthesis. If the dCDP pathway is traced from the common
pyrimidine precursor, UMP, it will proceed as follows: UMP ®
UDP ® UTP ® CTP
® CDP ® dCDP
® dCMP ® dUMP
® dTMP.
The synthesis of thymine nucleotides proceeds from other pyrimidine deoxyribonucleotides. Cells have no requirement for free thymine ribonucleotides and do not synthesize them. Small amounts of thymine ribonucleotides do occur in tRNA (an RNA species harboring a number of unusual nucleotides), but these Ts arise via methylation of U residues already incorporated into the tRNA. Both dUDP and dCDP can lead to formation of dUMP, the immediate precursor for dTMP synthesis (Figure 27.27). Interestingly, formation of dUMP from dUDP passes through dUTP, which is then cleaved by dUTPase, a pyrophosphatase that removes PPi from dUTP. The action of dUTPase prevents dUTP from serving as a substrate in DNA synthesis. An alternative route to dUMP formation starts with dCDP, which is dephosphorylated to dCMP and then deaminated by dCMP deaminase (Figure 27.28), leaving dUMP. dCMP deaminase provides a second point for allosteric regulation of dNTP synthesis; it is allosterically activated by dCTP and feedback-inhibited by dTTP. Of the four dNTPs, only dCTP does not interact with either of the regulatory sites on ribonucleotide reductase (Figure 27.26). Instead, it acts upon dCMP deaminase.
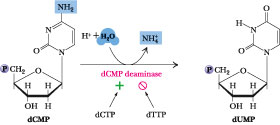 Figure
27.28 · The dCMP deaminase reaction.
Figure
27.28 · The dCMP deaminase reaction.
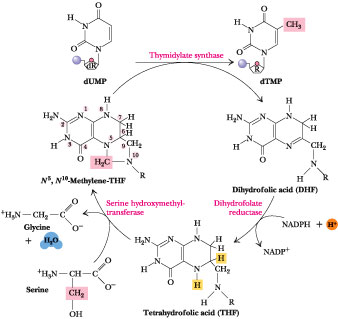 Figure
27.29 ·
The thymidylate synthase reaction. The 5-CH3 group is ultimately
derived from the b-carbon of serine.
Figure
27.29 ·
The thymidylate synthase reaction. The 5-CH3 group is ultimately
derived from the b-carbon of serine.
Synthesis of dTMP
from dUMP is catalyzed by thymidylate synthase (Figure 27.29). This
enzyme methylates dUMP at the 5-position to create dTMP; the methyl donor is the
one-carbon folic acid derivative N5,N10-methylene-THF.
The reaction is actually a reductive methylation in which the one-carbon unit is
transferred at the methylene level of reduction and then reduced to the methyl
level. The THF cofactor is oxidized at the expense of methylene reduction to
yield dihydrofolate, or DHF. Dihydrofolate reductase then reduces DHF back to
THF for service again as a one-carbon vehicle (see
Figure 18.35). Thymidylate synthase sits at a junction connecting dNTP
synthesis with folate metabolism. It has become a preferred target for
inhibitors designed to disrupt DNA synthesis. An indirect approach is to employ
folic acid precursors or analogs as antimetabolites of dTMP synthesis (Figure
27.30). Purine synthesis is affected as well because it is also dependent on THF
(Figure 27.3).
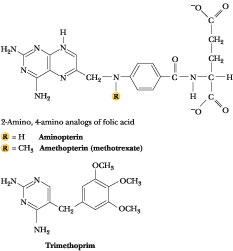
Figure 27.30 · Precursors and analogs of folic acid employed as antimetabolites: sulfonamides (see Figure 27.5), methotrexate, aminopterin, and trimethoprim. The latter three compounds bind to dihydrofolate reductase with about one thousand-fold greater affinity than DHF and thus act as virtually irreversible inhibitors.
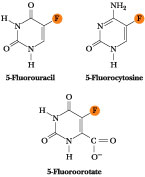 Figure
27.31 ·
The structures of 5-fluorouracil (5-FU), 5-fluorocytosine, and
5-fluoroorotate.
Figure
27.31 ·
The structures of 5-fluorouracil (5-FU), 5-fluorocytosine, and
5-fluoroorotate.
5-Fluorouracil (5-FU; Figure 27.31) is a
thymine analog. It is converted in vivo to 5'-fluorouridylate by a
PRPP-dependent phosphoribosyltransferase, and passes through the reactions of
dNTP synthesis, culminating ultimately as 2'-deoxy-5-fluorouridylic acid,
a potent inhibitor of dTMP synthase. 5-FU is used as a chemotherapeutic agent in
the treatment of cancer. Similarly, 5-fluorocytosine is used as an
antifungal drug because fungi, unlike mammals, can convert it to
2'-deoxy-5-fluorouridylate. Further, malarial parasites can use exogenous
orotate to make pyrimidines for nucleic acid synthesis whereas mammals cannot.
Thus, 5-fluoroorotate is an effective antimalarial drug because it is
selectively toxic to these parasites.