Research Notes
Transition Metal Hydrides as H· Donors
Tin hydrides (i.e., Bu3SnH) have traditionally been used as stoichiometric H donors in radical reactions. The byproducts from such tin hydrides are, however, toxic and difficult to remove, precluding the use of these reactions on industrial scale. We have shown that transition-metal hydrides (e.g., 1, 2, and 3) can be used as replacements for tin hydrides, and some can be used catalytically under H2, resulting in radical reactions that are green and atom-economical (generating almost no waste).
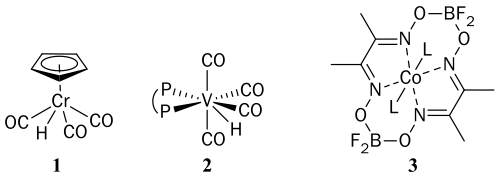
Transition-metal hydrides with reduced M-H bond strengths have enhanced reactivity, and can generate radicals directly from olefins by H· transfer. By measuring the physical properties of such metal hydrides (e.g., the strength of their M-H bonds, the rates at which they transfer H· to typical substrates, and the mechanisms of their reaction with H2), we can predict which hydrides will be effective catalysts for radical reactions.
M-H Bond Strength as a Determining Factor
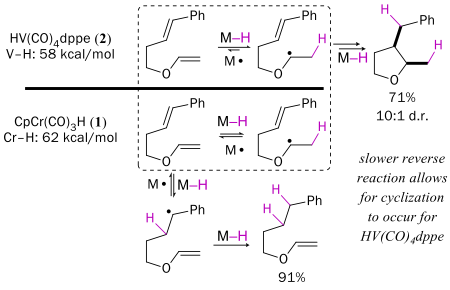
The Cr-H bond in CpCr(CO)3H is 62 kcal/mol, whereas the V-H bond in HV(CO)4dppe is only 56 kcal/mol. The difference in reactivity is large; for example, H· transfer to styrene is 10.6 x faster with vanadium. However, the V-H bond is so weak that V· does not reform the hydride by cleaving H2, so the vanadium system is not catalytic.
The M-H bond strength affects the outcome of radical cyclization reactions. For example, both CpCr(CO)3H and HV(CO)4dppe transfer H· to enol ethers, but the reverse reaction (transfer of H· back to the metal) is faster for Cr than for V. As a result alpha-alkoxy radicals generated from 2 cyclize, but those generated from 1 do not live long enough to do so.
The Reaction of Isonitriles with Group 4 Diene Complexes
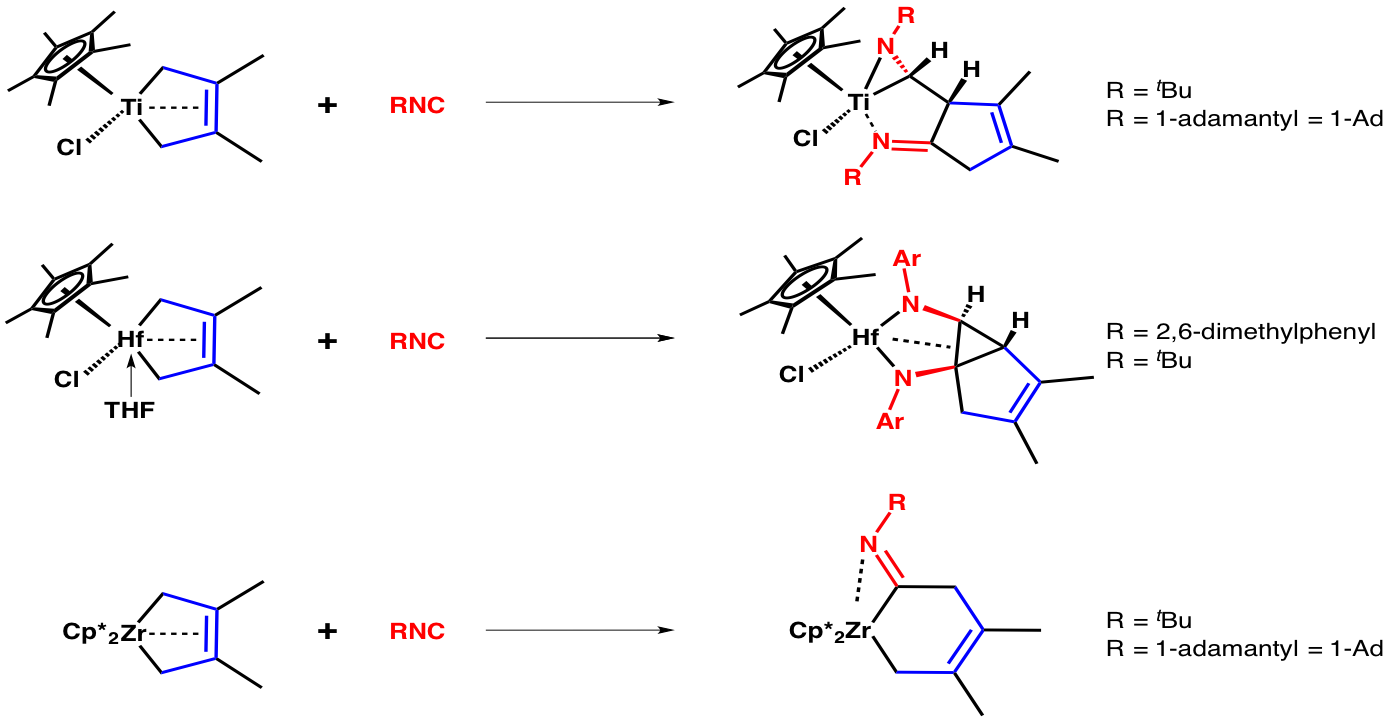
Organometallic chemistry is a powerful tool for coupling simple unsaturated molecules into complex structures. Isonitriles (RNC's) are isoelectronic with CO, and alternative sources of C1 in C-C coupling reactions. Changes on the N substituent in RNCs can effect insertion chemistry that is complementary to that of CO.
We have explored the reaction of isonitriles (RNC's) with Group 4 (Ti, Zr, and Hf) diene complexes. The nature of the insertion product changes with the metal and the isonitrile (RNC), yielding metallaaziridines (Ti), diazametallacyclopentanes (Hf), and eta2-iminoacyl complexes (Zr).
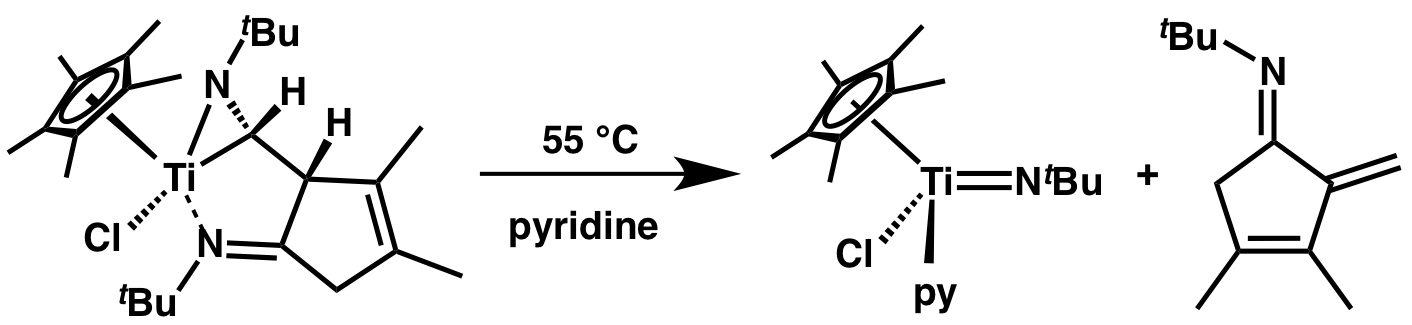
The titanium complex below fragments in the presence of pyridine to the known imido complex Cp*(Cl)Ti(NtBu)py and an alpha-methylene cyclopentenimine. The overall reaction is the first example of a formal [4 + 1] cycloaddition between an isonitrile and a diene.
The Interconversion of Metallaaziridine Enantiomers, and Their Use in Dynamic Kinetic Resolutions
eta2-Imine complexes of zirconium, or zirconaaziridines, have attracted attention as amino carbanion equivalents. Insertion of unsaturated organic compounds into the polar Zr-C bonds of zirconaaziridines leads to amines, allylic amines, heterocycles, diamines, amino alcohols, amino amides, amino amidines, and amino acid esters.
Zirconaaziridines contain a chiral center if their ring carbon bears two unequal substituents. That carbon is capable of configurational inversion if its eta2 isomer equilibrates with its eta1 analog (Scheme 1).
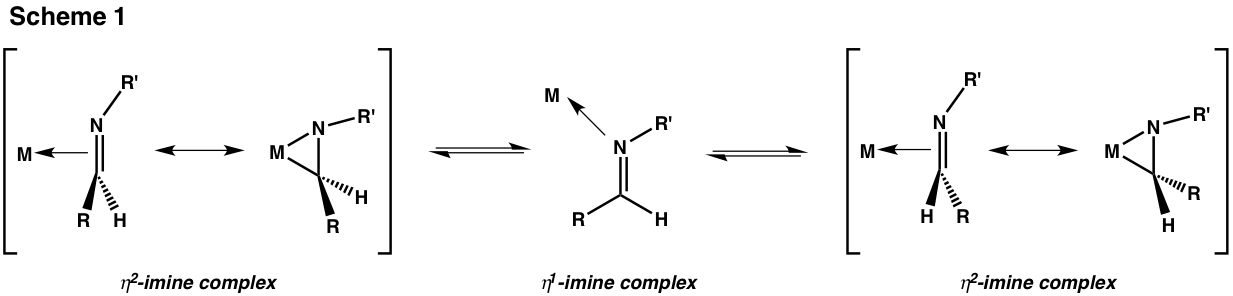
By treating a racemic zirconaaziridine 1 with a chiral electrophile (Scheme 2) under conditions where enantiomer interconversion is faster than electrophilic attack we have been able to achieve dynamic kinetic resolutions, i.e., to produce products enriched in one configuration of the carbon originally bound to Zr. We have found (R,R)-diphenylethylene carbonate ((R,R)-DPEC) to be an excellent chiral electrophile, giving zirconacycle 2 with de's up to 90%.
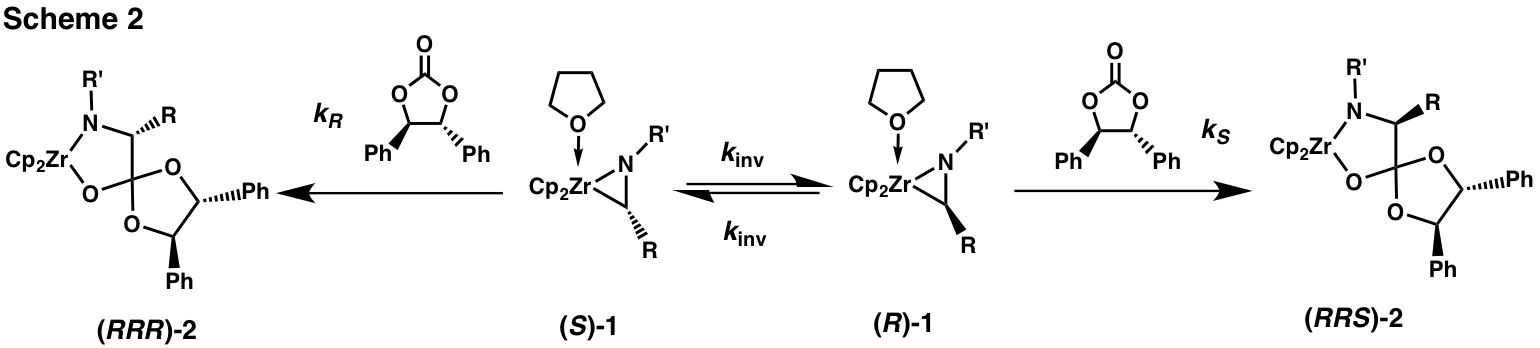
The stereochemistry of the new chiral center in 2 is determined by the competition between the (first order) interconversion of the enantiomers of 1 and the (second order) insertion of the carbonate into their Zr-C bonds (Scheme 2). When insertion is slow relative to the rate of enantiomer interconversion (the condition in eq 1, where Keq = 1) we will obtain a diastereomer ratio of kR/kS.
Achieving that maximum diastereomer ratio often requires slow addition, with a syringe pump, of the (R,R)-DPEC to the zirconaaziridine. This procedure fulfills the conditions of eq 1 by keeping [(R,R)-DPEC] low. However, for any given zirconaaziridine it is helpful to know the rate constant, kinv, for enantiomer interconversion. We have determined kinv in several cases, one fast (10 s 1), one slow (5.3 x 10-6 s 1) and one intermediate (9.1 x 10-3 s 1).
Motivated by the need for metallaaziridines with larger kinv, we have prepared a number of titanaaziridines, and have measured the rates by which their enantiomers interconvert by variable-temperature NMR.
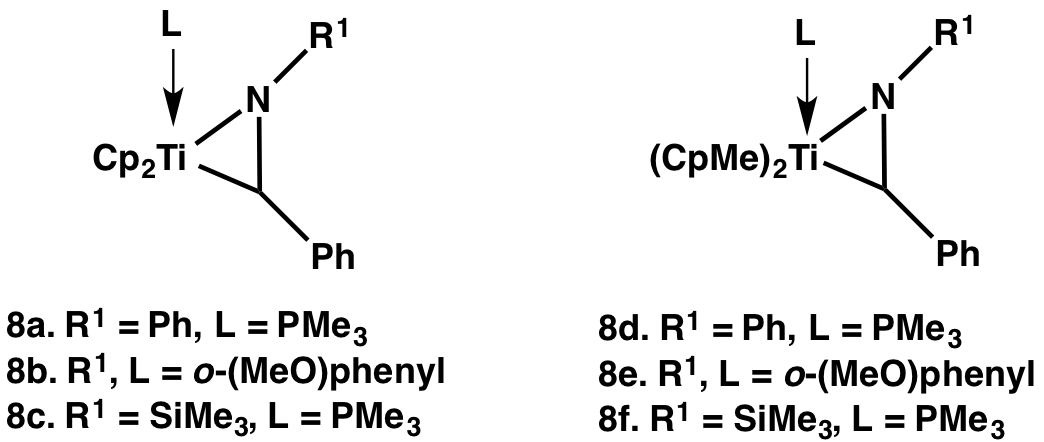
Methoxy-substituted titanaaziridines have the fastest enantiomer interconversion rate constants reported to date. For 3b we have used 1H NMR line broadening data to estimate deltaG+-298 = 11 kcal mol-1 (kinv 4 x 104 s-1 at 298 K). For the analogous zirconaaziridine, Cp2Zr(eta2-N(o-MeOPh)CHPh), we have reported kinv as 5.3 x 10-6 s-1 at 298 K and deltaG+-298 as 24.6 kcal mol-1. Titanaaziridine enantiomers thus interconvert orders of magnitude faster than the corresponding zirconaaziridine enantiomers, and dynamic kinetic resolutions should be more practical with titanium.
Polynuclear Copper Hydrides for the Generation of Electrons from Hydrogen
Polynuclear first-row hydrides are better "electron reservoirs" than mononuclear hydrides. We have observed a single-electron transfer as the initial step in the reaction of a copper hydride cluster [(Ph3P)CuH]6 with certain substrates (e.g., a pyridinium cation) in a stopped-flow apparatus. The inorganic product is a cation radical [(PPh3)CuH]6·+, which slowly evolves H2 and forms other polynuclear hydrides, for example [(PPh3)7Cu7H6]+.
We are trying to investigate the reactivity of the cation radical, and to regenerate the neutral [(Ph3P)CuH]6 with H2 in the presence of base. We hope to establish a copper-based catalytic system for the generation of electrons by H-2 oxidation.
We have also prepared copper hydride complexes of lower nuclearity with chelating phosphine ligands. For example, we have made a 48-electron trimer [(dppbz)-CuH]3 by use of the chelating ligand 1,2-bis(diphenylphosphino)benzene (dppbz). We are exploring its potential applications in catalyzing H2 oxidation.