3.1 THE ARTERIAL PRESSURE
A. MEASUREMENT OF ARTERIAL PRESSURE
The arterial pressure can be determined directly by introducing into a systematic artery a needle or a catheter that is connected to a pressure measuring device. The pressure measuring device may be a pressure transducer connected to an amplifier-recorder system which allows the recording of phasic changes in arterial pressure with each cardiac cycle, or it may be a mercury manometer which gives only the mean arterial pressure due to damping by the mercury. The direct method is used in investigations on experimental animals and clinically in intensive care units, in the operating room, and in the cardiac catheterization laboratory. When direct pressure measurement is made by introducing the needle in a direction perpendicular to the flow, one measures the lateral pressure exerted radially on the vessel wall, i.e., the internal pressure which provides the transmural pressure. If the needle or the catheter is pointed in a direction against the flow, then the end pressure measured also includes an additional pressure component due to the conversion of the kinetic energy. In the systemic arteries, however, the kinetic component is generally small, and the end pressure is not appreciably higher than the lateral pressure.
Routine determination of arterial pressure in human subjects or patients is performed by an indirect method using a sphygmomanometer. In this method, an inflatable cuff is wrapped around the upper arm, and a stethoscope head is pressed gently against the antecubital fossa for auscultation of sounds in the brachial artery. Before inflation of the cuff, blood flow through the artery is laminar and therefore, there is no audible sound. When the cuff pressure is raised above the systolic pressure, there is total cessation of flow through the arm and again there is no sound. As the cuff is gradually deflated, a sharp, tapping sound can be heard when the cuff pressure is reduced to the level of the systolic pressure, thus allowing the occluded artery to open briefly. Therefore, the reading of the manometer at the first occurrence of this sound corresponds to the systolic pressure. With progressive reduction in cuff pressure, the sound recurs with each systolic peak, but the tone becomes softer and the volume louder, as the arterial opening becomes wider and stays open longer. It has been found by comparison with the direct pressure recording that the sound becomes muffled when the cuff pressure equals the diastolic pressure. A further reduction of the cuff pressure to approximately 5 mmHg below the diastolic pressure causes complete disappearance of sound. Instead of using the auscultatory method, the systolic pressure can also be estimated, with lesser accuracy, by observing the onset of oscillation of the mercury column in the manometer or by palpating for the onset of a pulse in the radial artery during cuff deflation.
B. THE MEAN ARTERIAL PRESSURE
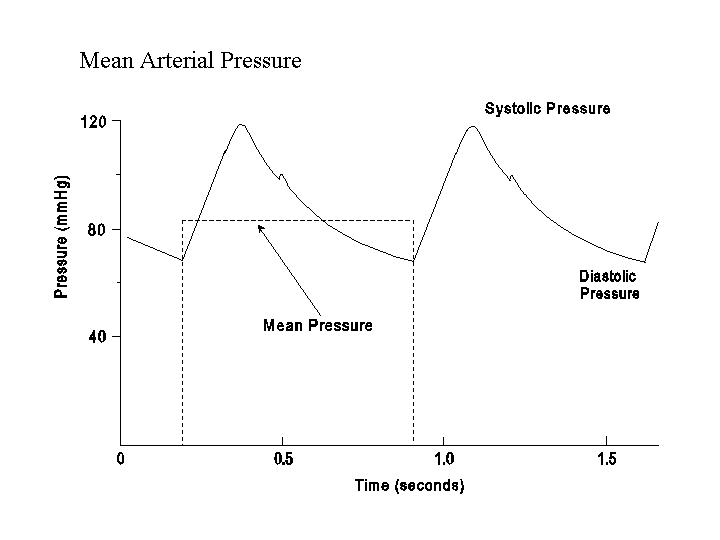
The arterial pressure varies in a pulsatile fashion between the systolic and diastolic values. The mean arterial pressure is the mean value obtained by integrating the pressure over time, i.e., with equal weight given to each infinitesimal time interval. Therefore, the mean arterial pressure is not equal to the simple average of the systolic and diastolic pressures. In direct measurements of arterial pressure, the mean pressure can be obtained by graphic integration of the pressure tracing, by electronic integration in an amplifier-recorder system, or by mechanical damping in a mercury manometer. As a rule of thumb, the mean arterial pressure (Pm) can be estimated from the systolic pressure (Ps) and diastolic pressure (Pd) obtained in indirect measurements by the following equation:
The greater weighting toward Pd reflects the fact that the arterial pressure is closer to the Pd for longer periods than it is to Ps (see Figure).
C. PRESSURE CHANGE ALONG THE ARTERIAL TREE
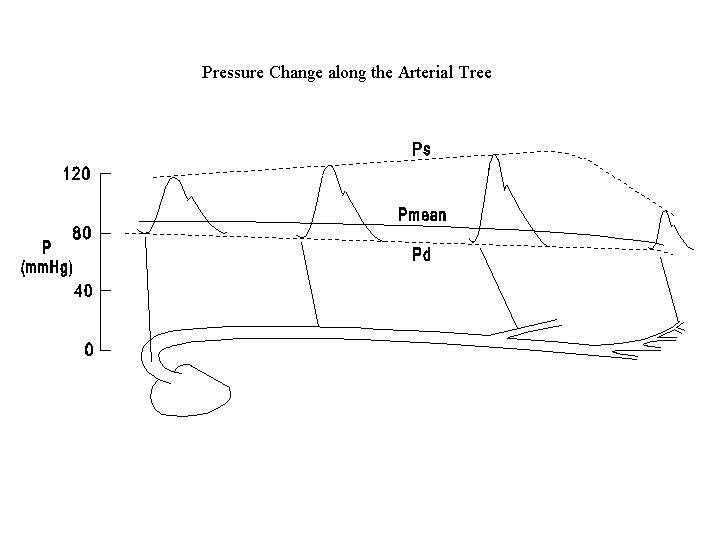
The mean arterial pressure decreases progressively from the aorta toward the peripheral branches, reflecting the energy dissipation in flow. In comparison to the root of the aorta the large and medium-sized arteries have a slightly lower mean pressure, but they have a slightly higher systolic pressure. The higher systolic pressure in the large and medium arteries is best explained by the reflection of pressure wave from the small arteries. With each systole, the arterial wall propagate the pressure wave peripherally at a rate (approximately 500 cm/sec) much faster than the rate of blood blow (approximately 20 cm/sec). The pressure wave cannot be propagated forward through the high resistance small arteries and is reflected to travel in a retrograde fashion. As a result of the summation of the reflected wave with the forward wave, the systolic pressure in the more centrally located medium-sized arteries is enhanced. The diastolic pressure shows a continuous decrease from the aorta toward the periphery. Therefore, the pulse pressure, which is the difference between systolic and diastolic pressure, gradually increases from the aorta toward the medium-sized arteries.
D. INFLUENCE OF ARTERIAL CAPACITANCE ON ARTERIAL PRESSURE
The capacitance of the arterial tree and the peripheral resistance to flow cause the storage of a portion of the systolic ejection in the arterial system followed by a gradual release during diastole. Therefore, the normal capacitance of the arterial wall serves to minimize the rise of pressure during systole as well as the fall of pressure during diastole. A decrease in arterial capacitance, therefore, would cause an increase in systolic pressure and a decrease in diastolic pressure, leading to a widening of the pulse pressure without changing the mean arterial pressure. If the decrease in distensibility also involves the small arteries and other resistance vessels, then the resulting increase in flow resistance will raise the systolic, diastolic, and mean pressures, as seen in the case of aging.
E. FACTORS INFLUENCING THE MEAN ARTERIAL PRESSURE
The pressure drop between two points is equal to the product of flow and resistance. In the systemic circulation, the driving pressure is that in the arterial system and the end-point of the circuit is the right atrium, which has a pressure near zero (i.e., atmospheric). Therefore,
where the total peripheral resistance (TPR) is the sum total of all resistances in the systemic circulation. The factors regulating cardiac output and TPR will be discussed in the subsequent sections 3.2 and 3.3, respectively.
Cardiac output is equal to the product of heart rate and stroke volume, which in turn is equal to the difference between the end-diastolic volume (EDV) and the end-systolic volume (ESV) of the ventricle.
In each cardiac cycle, systolic ejection causes the ventricular volume to be reduced from EDV to ESV, and diastolic filling causes the ventricular volume to rise from ESV to EDV. Regulatory mechanisms exist for the control of both systolic ejection and diastolic filling, and these two processes are interrelated to maintain their equality in a steady state.
A. REGULATIONS OF HEART RATE
The factors controlling heart rate are discussed in Chapter 1. Cardiac sympathetic nerve impulses and catecholamines cause an acceleration of heart rate. Vagal impulses, on the other hand, cause cardiac slowing.
B. REGULATION OF SYSTOLIC EJECTION
The force generated by the ventricular myocardium and the volume and rate of ejection depend on the external working conditions as well as the intrinsic contractility. The intrinsic contractility of the myocardium is enhanced by sympathetic nerve impulses and catecholamines. The influences of external working conditions serve to adjust the systolic ejection in response to alterations in peripheral circulatory dynamics. Thus, the increase in force of myocardial contraction consequent to an increase in EDV (Starling�s law) allows an adjustment of ventricular output in accordance with variations in return from the venous system. The reduction in the rate of systolic ejection following elevation of aortic pressure leads to changes in cardiac output opposite to alteration in peripheral resistance, thus tending to maintain a relatively constant arterial pressure.
C. REGULATION OF DIASTOLIC FILLING
The volume of blood filling the ventricle from ESV to EDV in each cycle is a function of the duration of filling (diastolic interval) and the rate of filling (venous return).
(a) Diastolic interval.
The diastolic interval is curtailed by an increase in the heart rate, especially when the heart rate is faster than 100 beats per min, and this leads to a decrease in diastolic filling (decrease of EDV). When cardioacceleration is brought about by sympathetic stimulation, however, there is generally a concomitant stimulation of myocardial contractility, which enhances systolic ejection (decrease of ESV). Therefore, when the cardiac sympathetic nerves are activated, e.g., during muscular exercise, the decreases of both EDV and ESV result in a stroke volume which is essentially unchanged or slightly increased, and the cardiac output increases mainly due to the acceleration in heart rate.
(b) Relation between blood and vascular capacity.
The rate of diastolic filling via venous return is determined by the fullness of circulatory system, the right atrial pressure, and the resistance in the venous return path. The fullness of the circulation refers to the relation between blood volume and vascular capacity, especially that of the veins. For a given level of vascular capacity, the fullness of the circulation and the rate of venous return vary directly with the volume of blood. For a given blood volume, the fullness of circulation varies inversely with vascular capacity. That is, an increase in venous capacity would raise the proportion of blood volume trapped in the peripheral veins and reduce the venous return. Sympathetic nerve impulses and catecholamines cause a reduction in venous capacity by causing contraction of venous smooth muscle thus facilitating venous return. The degree of venous distention is affected not only by the tension in the venous smooth muscle, but also by mechanical factors originating extravascularly and intravascularly. The influence of the extravascular factor can be seen in the venous return through skeletal muscle undergoing exercise. Veins in the leg contain valves which permit only centripetal flow. When the leg muscles contract, the compression on the vein causes blood to flow out of the compressed segment towards the heart. When the leg muscles relax, the decompression of the vein allows blood to enter this segment from the more distal segments. Thus, rhythmic exercise of the leg muscle serves as muscle pump to facilitate venous return to the right atrium. Similarly, rhythmic change in the intrathoracic pressure with respiratory cycle functions as a respiratory pump.
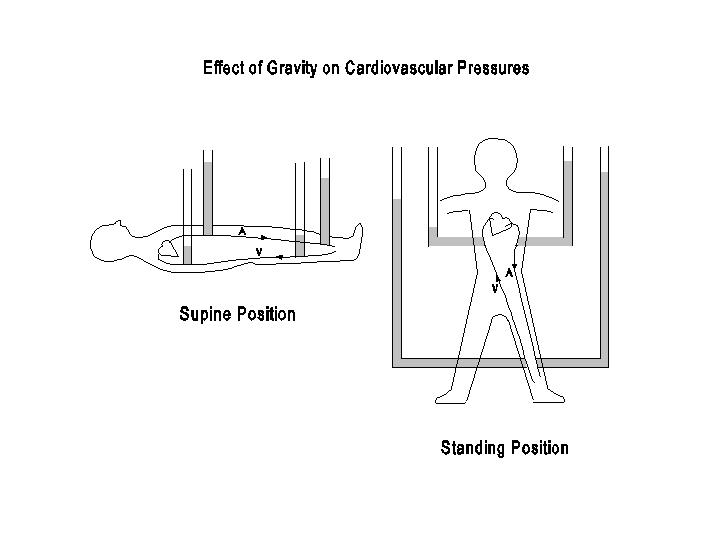
(c) Effect of gravity on venous return.
The influence of intravascular pressure on venous distension can be illustrated by the effect of gravity. When a person lies down in a horizontal position, there is negligible hydrostatic effect due to gravity on the various parts of the circulation, since all parts are at nearly the same hydrostatic level as the heart. When the person assumes a standing position, however, the hydrostatic effect becomes significant at locations distant from the heart. This is most pronounced in the feet. Therefore, the pressure measured in each vessel of the foot includes not only the pressure generated by the pumping on the heart, but also an additional component due to the hydrostatic pressure exerted by the column of the blood between the right atrium and the foot. This hydrostatic pressure varies with height and is approximately 100 mmHg. Therefore, upon assumption of the erect position, the mean arterial pressure on the foot may rise from 90 to 190 mmHg and the venous pressure from vessel 10 to 110 mmHg. If the vessels were made of rigid tubes, such hydrostatic influences would have had no effect on venous return, as in siphoning. Because the vessels are distensible, however, the additional hydrostatic pressure raises the transmural pressure and increases the blood volume contained in the vessel. This effect is especially pronounced in the veins, due to their greater capacitance than the arteries. As a negative hydrostatic pressure exists in the vessels above the heart, e.g., the carotid arteries, a decrease in transmural pressure occurs there upon standing. As is discussed in Chapter 4, a decrease of transmural pressure in the carotid region leads to a reflex stimulation of the sympathetic system which then causes a decrease of venous capacitance and a reduction of hydrostatic pooling. The negative hydrostatic pressure in the cerebral vessels upon standing does not lead to vascular collapse, because of the existence of the rigid cranium.
(d) The right atrial pressure.
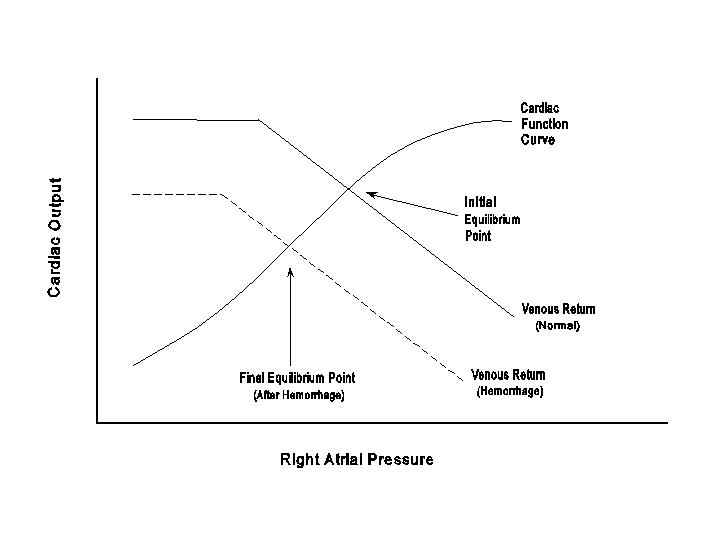
The right atrial pressure is normally near the atmospheric level. When there is a decrease in atrial capacitance (e.g., pericardial fluid accumulation) or a reduction in right ventricular output (e.g., right ventricular failure), the resulting elevation of right atrial pressure causes a decrease in systemic venous return. It should be noted, however, that an elevation of right atrial pressure can also result from an increase in systemic venous return. Under that circumstance, the increased atrial pressure serves to increase ventricular output through the increase in diastolic filling (Starling�s law). The above discussions indicate that the atrial pressure reflects a balance between cardiac output and venous return, and hence it has a pivotal significance in circulatory dynamics. Through its influence on the right atrial pressure, an alteration in either the cardiac output or the venous return can cause maintaining the balance between these two flow parameters and the continuity of circulation. For example, with hemorrhage, right atrial pressure decreases and leads to a reduction in venous return. This reduction in right atrial pressure in turn leads to a reduction in cardiac output due to Starling�s law. The new, reduced steady-state right atrial pressure after hemorrhage satisfies both the venous return-venous pressure and the cardiac output-venous pressure relations (see Figure).
(e) Resistance to venous return.
The resistance to venous return is a function of vascular geometry, especially that of the venous side of the circulation, and blood viscosity. A discussion of factors affecting these parameters is given in the next section on total peripheral resistance. A decrease in the size of the veins may reduce the capacitance and increase the resistance, and these two effects have opposite influences on venous return. As a rule, moderate increases in the tension of venous smooth muscle by sympathetic stimulation primarily decreases the capacitance without a marked increase in resistance, thus facilitating the venous return. On the other hand, a severe constriction of the venous lumen, especially when a local segment of a large vein is affected, the main effect would be an increase in resistance and a reduction in venous return. The facilitation of venous return by a low blood viscosity occurs in patients with anemia. The factors regulating cardiac output are summarized on the next page together with those controlling peripheral resistance.
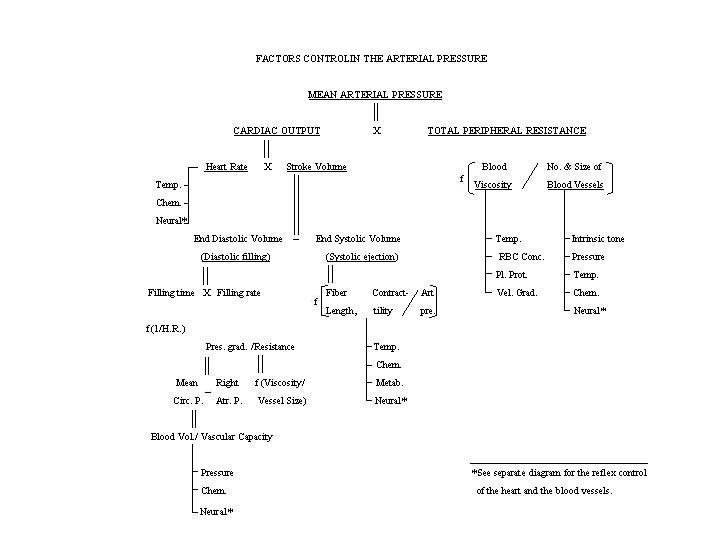
Resistance to flow is determined by blood viscosity and vascular hindrance, which varies inversely with the number and size of blood vessels.
The lengths of the vessels do not change significantly except during growth. The factors regulating peripheral resistance are summarized in the diagram above (together with factors controlling cardiac output) and discussed below.
A. BLOOD VISCOSITY
The viscosity of blood, as that of most liquids, varies inversely with temperature. Exposure to cold temperature causes an increase in blood viscosity, mainly during flow through the cutaneous circulation.
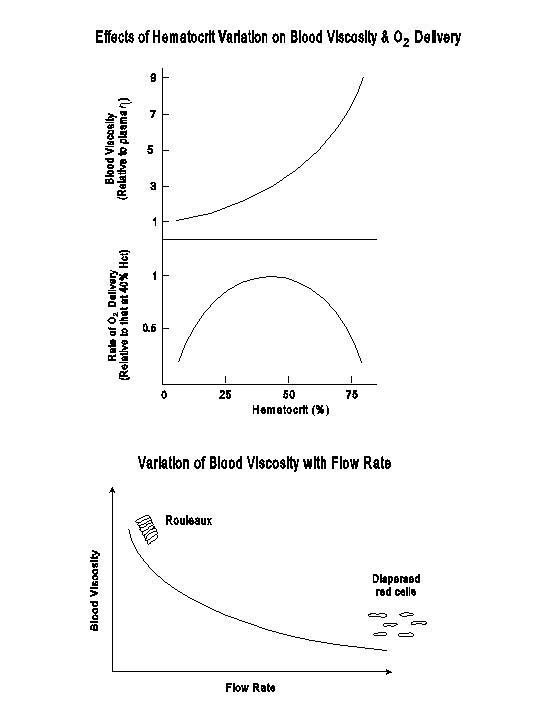
The concentration of erythrocytes in the blood has a strong influence on blood viscosity. At a hematocrit of 40-45%, blood viscosity is approximately 3 times the value for plasma and approximately 5 times that of water. Blood viscosity shows a curvilinear relation with the hematocrit and it increases sharply when the hematocrit is raised much beyond the normal range (see following diagram, upper panel). This increase in viscosity with cell concentration would have even much steeper if the red cells had been rigid. The remarkable deformability of the normal red cells, besides making it possible for them to traverse narrow capillaries, serves to minimize the rise of blood viscosity with increasing cell concentration. The relation between blood viscosity and red cell concentration indicates that, when all other conditions are equal, blood flow would decrease with an increase in hematocrit, especially at high hematocrit levels. When respiratory functions are normal, the arterial oxygen content is directly proportional to the hamatocrit. Therefore, the rate of oxygen delivery to tissues, which is a product of blood flow and arterial oxygen content, shows a bell-shaped relation with the hematocrit value (lower panel of the diagram on the effect of hematocrit variation). Oxygen delivery is slow at low hematocrit levels because of the low arterial content, and it is slow also at high hematocrit levels due to the high viscosity. The optimum hematocrit for maximum oxygen delivery is approximately 40%, i.e., about the normal value.
An elevation of the concentration of plasma proteins, especially fibrinogen and globulins, causes an increase in blood viscosity by two mechanisms. First, the plasma viscosity is raised. Second, when the flow rate is slow, fibrinogen and globulins can bridge red cell surfaces to form rouleaux, and such red cell aggregation also causes an increase in blood viscosity. High flow rates are associated with a high mechanical shearing force which tends to disperse the aggregates and align the deformed cells, thus lowering the blood viscosity.
Blood viscosity is independent of the size of the blood vessel when the diameter is above approximately 0.5 mm. A decrease of vessel diameter below this value causes a progressive reduction in blood viscosity, and a major reason is that the blood entering these small vessels tend to have a lower hematocrit than that in the large vessels.
B. VASCULAR HINDRANCE
Alterations in blood viscosity usually do not occur very suddenly. In contrast, the number and size of blood vessels can change more rapidly to effect the vascular hindrance. The major resistance vessels are the arterioles and small arteries, where the pressure drop is the steepest. These vessels have a rich content of smooth muscle which has an intrinsic tone and can generate significant active stress in response to physicochemical or neurohumoral stimuli. Furthermore, the small ratio for the lumen radius to wall thickness in the resistance vessels can amplify the vasoconstriction due to smooth muscle contraction.
(a) Physical factors affecting vascular hindrance.
A moderate decrease in temperature (e.g., 10°C) causes vasoconstriction and a moderate increase in temperature (e.g., 35°C) causes vasodilation. Extreme changes in temperature (e.g., 0°C or 45°C) cause vasodilation, an effect similar to that induced by other noxious stimuli resulting from mechanical stretching or chemical irritation (see discussion below in relation to stretching). These direct effects of temperature on vessel size are important mainly in the cutaneous circulation, whereas the core temperature in the body is normally regulated within narrow limits.
The size of blood vessels may be altered in response to mechanical factors originating intravascularly (pressure changes) or extravascularly (compression or stretching). The circumferentially arranged smooth muscle fibers and other fibers in the vessel wall are extended by an increase in the transmural pressure difference or by external stretching forces. The changes in vessel diameters resulting from the stretch vary in different circulatory beds. Some vessels show mainly a passive response to stretch, and an increase in transmural pressure causes vasodilation. Other vessels, e.g., the resistance vessels in the renal and cerebral circulations, however react to mechanical stretch by an active contraction of the smooth muscle. As a result, an increase in arterial pressure can lead to a vasoconstriction and an increase in resistance. Since blood flow is determined by the pressure/resistance ratio, the active response of vascular smooth muscle to stretch serves to maintain a relative constancy of flow in the face of alterations in arterial pressure. This process is referred to as autoregulation of flow by mechanical factors, as it does not involve any neural or hormonal mechanisms. The above discussions on passive and active responses of resistance vessels apply to their reactions to moderate mechanical stimuli. Strong mechanical stimuli exert effects similar to that induced by other noxious stimuli, such as extreme thermal changes or histamine release. If a firm stroke is drawn over the skin with a blunt instrument, there results a characteristic triple response, which consists of a red line on the path stroked, a reddish flare surrounding the red line with an irregular contour, and a wheal in the same area. The red line results from the dilation of minute vessels (e.g., metarterioles and precapillary sphincters) due to the local release of histamine and/or bradykinin. The mechanical irritations and/or the chemical agents released cause a stimulation of some sensory endings located in the line of stroke. These sensory impulses are conducted not only along the main sensory nerve toward the dorsal root and central nervous system, but also along any collateral branches arising from the main nerve path.
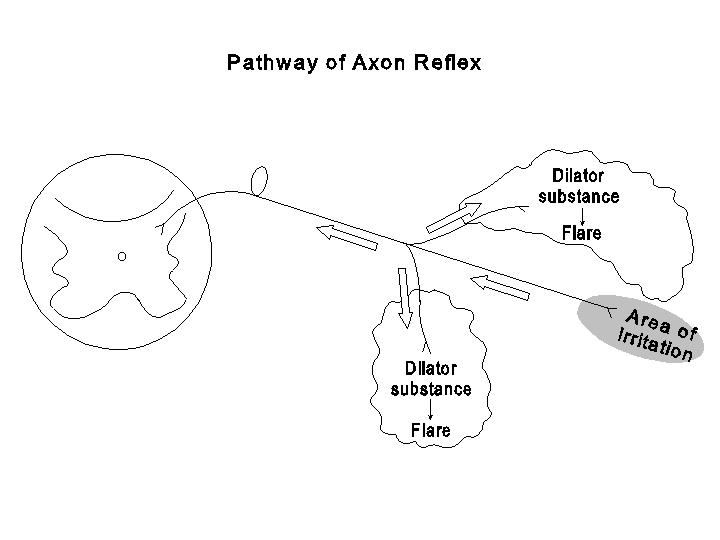
A vasodilator substance, probably adenosine triphosphate (ATP), is then released from the terminals of these collateral branches (dorsal root vasodilator fibers) and cause the flare. Therefore, the flare is the result of a reflex-like neural mechanisms called axon reflex, which involves only branches of the same axon. The irregular contour of the flare outlines the area of capillaries being filled with blood to the dilation of feeding arterioles resulting from the axon reflex. Histamine and bradykinin cause an increase in capillary permeability and an escape of plasma proteins and fluid into the extravascular (interstitial) space. This process is facilitated by the increase in capillary pressure due to the dilation of precapillary vessels. The accumulation of such edema fluid in the interstitial space is the basis for wheal formation.
(b) Chemical factors affecting vascular hindrance.
Histamine is present in the mast cells in various tissues and is released in inflammatory processes, allergic responses and antigen-antibody reactions. Localized release of histamine due to noxious stimuli causes triple response in the area stimulated. Generalized release of large amounts of histamine, e.g., in anaphylactic shock due to serum sickness, may cause severe vasodilation and precipitous drop in arterial pressure.
Sweat glands and salivary glands can secrete kallikrein, which is an enzyme acting on the α2-globulin in plasma or interstitial fluid to produce a vasoactive polypeptide, bradykinin. Bradikinin causes vasodilation and leads to an increase in capillary permeability.
The kidney secretes the enzyme renin response to a decrease in renal blood flow and other factors. Renin can act on the plasma α2-globulin to produce the decapeptide angiotensin I, which does not have significant physiological activities. Angiotensin I is converted by the angiotensin converting enzyme in the lung to angiotensin II, an octapeptide which causes constriction of arterioles in the systemic circulation. Angiotensin II also stimulates the adrenal cortex to secrete aldosterone, which causes Na retention and an expansion of extracellular fluid volume, including the plasma volume. Therefore, overactivity of the renin-angiotensin system can increase the cardiac output via blood volume expansion and raise the peripheral resistance by arteriolar constriction. These pathophysiological changes may have considerable significance in the pathogenesis of some forms of hypertension.
Another vasoactive polypeptide is vasopressin (or anti-diuretic hormone, ADH), which is an octapeptide secreted by the supraoptic nuclei of the hypothalamus and stored in the neurohypophysis. Although the primary action of vasopressin is to reduce urine flow by acting on the renal tubules, it may also cause constriction of arterioles. It has been shown that vasopressin can act synergistically with cathecolamines and enhance their vasoconstricting effect.
Prostaglandins (PGs) are 20-carbon, hydroxy fatty acids biosynthesized from arachidonic acid. A microsomal enzyme PG synthetase causes the formation of unstable PG endoperoxides PGG2 or PGH2 which are transformed to (a) prostacyclin (PGI2) by prostacyclin synthetase, (b) thromboxane A2 (TXA2) by thromboxane synthetase, or (c) PGE2, PGF2 or PGD2, PGI2 is a potent vasodilator and an inhibitor of platelet aggregation. TXA2 can induce platelet aggregation and vasoconstriction in vitro. PGE2 is a vasodilator, while PGF2 can cause vasoconstriction. The PGs are ubiquitous substances which can be formed in almost all organs in response to appropriate stimuli, and they may mediate or modulate the vasoactive actions of many other humoral agents.
The influence of catecholamines on vascular resistance has been discussed in the sections on the Autonomic Nervous System.
The metabolic environment (the partial pressures of O2 and CO2 and the pH) in various tissues depends on the balance between circulatory transport and tissue metabolism. Whenever circulatory transport is insufficient to meet metabolic needs, there would be a reduction in tissue pO2, an increase in pCO2, and a decrease in pH. These metabolic changes cause vasodilation, though the relative importance of these changes varies with the organs or tissues in question. As a result of the vasodilation and decrease in resistance, blood flow through the region rises to minimize the tissue metabolic changes. By such a mechanism of autoregulation by metabolic factors, the rate of blood flow and the metabolic environment of tissues can be regulated locally independent of neural or hormonal influences.
(c) Neural regulation of vascular hindrance.
Dorsal Root Vasodilator Fibers:
As discussed above, the axon reflex initiated by noxious stimuli leads to vasodilation of the areas immediately surrounding the region
affected. These changes are usually localized and exert no significant influence on the total peripheral resistance.
Parasympathetic Fibers:
The parasympathetic fibers release acetylcholine at their nerve endings to cause vasodilation. Since these parasympathetic
cholinergic fibers have a rather limited distribution (mainly to the facial and pelvic regions), they do not contribute
greatly to the overall regulation of the total peripheral resistance.
Sympathetic Cholinergic Fibers:
Some of the sympathetic postganglionic fibers innervating the blood vessels in the skeletal muscle release acetylcholine
which causes vasodilation. These sympathetic cholinergic impulses are elicited by emotional factors and anticipation of
exercise, and they are not activated in most other circumstances.
Sympathetic Adrenergic Fibers:
Most sympathetic postganglionic fibers innervating the vascular smooth muscles are adrenergic, and the norepinepherine
released at these nerve endings causes vasoconstriction except in the cerebral and coronary circulation. These sympathetic
adrenergic fibers constitute the major efferent for the control of vascular
hindrance (see next Section).