Biomedical Engineering Laboratory IV
Optical Microscopy Laboratory
Professor James Thomas
Spring 1999
Introduction
Optical microscopy has enjoyed a renaissance in biological and biomedical applications. Although using visible light (as opposed to electrons or x-rays) limits resolution to approximately one-quarter micron, optical microscopy can be routinely used to study living cells. In addition, it is important to understand that although resolution is limited, optical microscopy has essentially unlimited precision, provided one can collect enough light. This realization has allowed scientists to track intracellular organelles or marker beads to an accuracy of less than a nanometer, and to measure the viscoelastic and mechanical properties of the cellular cytoplasm, auditory hair cell bundles, etc.
An important advance that was critical for the modern use of optical microscopy in cell biology was the development of phase contrast techniques. The first, and still most popular technique, was developed by the Dutch physicist Fritz Zernike in 1934, and is called Zernike Phase Contrast, or simply phase contrast. Other techniques, such as Differential Interference Contrast (DIC) and Hoffman Modulation Contrast, are derivatives of Zernike's original idea. Zernike was awarded the Nobel Prize for his invention in 1953.
In conventional ("bright field") microscopy, an enlarged image of the sample is formed by the microscope objective, and this image is then viewed using the eyepiece. (The role of the eyepiece is simply to allow you to inspect the image formed by the objective at a very close distance, just as a magnifying lens will allow you to put your eye very close to a small object and still focus. More on this later.) In conventional microscopy, if the sample does not absorb light, there will be essentially no contrast in the visible image (it will be all white). Most living cells absorb very little light (exceptions: red blood cells, chloroplasts, melanoma cells) and are consequently difficult to see in bright field microscopy. Although the cells absorb very little light, they do have different thicknesses and different refractive indices in their different parts, which leads to phase differences in the light that passes through them. The phase of a beam of light is generally unobservable (we just see the intensity, not the phase), but Zernike figured out a way to make phase differences appear in the image as intensity differences.
Critical Concepts
The following concepts are critical to understanding optical microscopy, as well as most other imaging modalities. You are expected to understand them, and will be tested on them on the final exam.
Focal length
Magnification (by the objective, and by the eyepiece)
Fourier optics
Numerical aperture
Resolution Precision Back Focal Plane
Front Focal Plane
Phase Ring
Phasor Diagram
Chromatic aberration
Spherical aberration
Laboratory Exercises
As engineers, it is important that you understand not only what biomedical instruments can do, but also how they do what they do. This will help you to find new applications for existing instruments and to design new instruments when necessary. To help you understand the "black box" that is the optical microscope, we have removed the box, leaving only the optical elements mounted on an optical rail. These elements consist of
Ray Optics
The fundamental equation of ray optics is the lensmaker's equation,
![]()
where i is the distance to the image, o is the distance to the object, and f is the focal length. If an object is infinitely far away from the lens, an image will be formed in the focal plane, a plane a distance f from the lens. Rays of light are bent by refraction as they move through the lens, but for an ideal (infinitely thin) lens, the ray that passes through the center of the lens is not deflected. To show how light from an object travels through a lens, it is customary to draw two rays from the tip of the object (usually represented schematically as an arrow). One ray is drawn through the center of the lens, and the other is drawn parallel to the optical axis. On the other side of the lens, if it is a converging lens, the first ray is undeflected, and the second will be deflected through the focal point. Where the two rays intersect, the image will be formed.
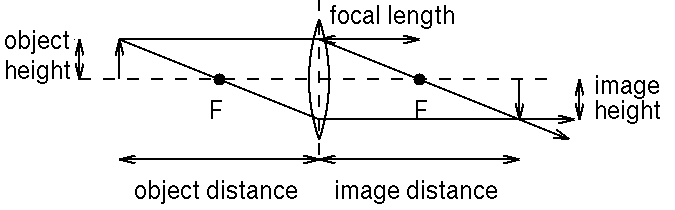
Figure 1. Conventional ray drawing of a lens making an image of an object. The dotted line is the optical axis.
Q. What would happen if the object were closer to the lens than f?
If the image is larger than the object, we say that there is magnification. The magnification is the ratio of the size of the image to that of the object. Note that, since the center ray is undeflected, the ratio of image size to object size will be the same as the ratio of image distance to object distance. (Why?)
Aberrations
Lenses are ground with spherical surfaces. Therefore, rays that are far away from the optical axis do not focus at the same point as rays near the axis. This is known as spherical aberration. In a microscope objective, it will cause the center of a flat field to focus at a different place from the periphery. It can be corrected by using combinations of lenses made from different glass (which will have different indices of refraction.) An objective that is corrected to have a flat field is called a PLAN objective.
In addition, the index of refraction of glass varies with wavelength, being generally higher for shorter wavelengths. This means that a given lens will deflect blue light more than red, i.e. that the focal length for blue light is shorter than that for red. Consequently, the blue light from an object will focus at a different place than the red light. The solution is again to use multiple lenses of different glasses. In this way, it is possible to design a lens that has the same focal length at two colors (red and blue, for example), which is called an achromat lens. Note that an achromat will not have the same focal length for colors in between blue and red! A three-lens system can be designed that has the same focal length for three colors, and is termed an apochromat.
Laboratory Task 1
Position the condenser so as to illuminate a rule, or even a slide with marks on it, held in the sample holder. Use a piece of paper to find where the image of the rule is. By making measurements of the distance between the lens, the sample, and the image, estimate the focal length of the objective lens. What are the quantities you should plot to get the best estimate?
What is the magnification of the 10X objective? If you require the image to be exactly 16 cm away from the objective, what is the magnification?
The Eyepiece
The role of the objective is to make an image of the sample approximately 10-20 cm away. This image is viewed up close using the eyepiece. Can you see the image without using an eyepiece? The eyepiece takes the intermediate image and makes a second image that is at approximately 25 cm away from the eye. The second image is a virtual image, unlike the image made by the objective. A real image can be seen if a piece of paper is placed at the position of the image, but a virtual image is just the location that the rays of light appear to be emanating from. Is the distance between the eyepiece and the intermediate image larger or smaller than the focal length of the eyepiece? The magnification of the eyepiece is figured in the same way as for the objective _ it is just the ratio of the size of the virtual image to the intermediate (real) image. The virtual image is assumed to be at 25 cm. If an eyepiece has a stated magnification of 10X, how far away from the intermediate image should it be located?
The Focal Planes
The objective forms an image of the sample some 16 cm away. Consequently, the sample must be very close to the focal plane of the objective. This focal plane can also be called the "front focal plane". The objective makes an image of the front focal plane 16 cm into the microscope, and this plane is called a "conjugate front focal plane". The condenser can also make an image of the sample, farther away. This other plane would also be called a "conjugate front focal plane."
Laboratory Task 2
Use an auxiliary light source, projected back through the sample, to find the conjugate focal plane behind the condenser. Now, without moving the condenser, reinsert the objective and eyepiece, and focus on the sample. If you now take a piece of paper and partially block the beam at the conjugate front focal plane, what do you see?
While it is important to be able to focus on the sample, it is sometimes important to defocus an object as much as possible. For example, the filament of the lamp should be completely defocused in the sample plane _ otherwise, there will be brightness variation across the sample. To be "completely defocused" means that the filament should be "at infinity." When an object is at infinity, it will focus at a focal plane _ but now this focal plane is on the other side of the objective lens. This focal plane is called the "back focal plane". There is a special tool for looking at the back focal plane in a microscope objective, an eyepiece that is called a telescope eyepiece.
Laboratory Task 3
Use the telescope eyepiece to look at the back focal plane of the 10X objective. What do you see? Focus the condenser until the filament is focussed in the back focal plane. This is called Kóhler illumination.
Diffraction
Ray optics is sufficient to understand the concepts of the focal planes and magnification, but it cannot help us to understand resolution. Microscopy of any kind is limited by the wave nature of the probing particles. Particles of visible light, or photons, have wavelengths of about a half a micron; particles of electrons have wavelengths that are thousands of times shorter. (In fact, the resolution in an electron microscope is limited by the difficulty of making good electron lenses, not the wave nature of the electron itself.)
How do we understand waves? The fundamental principle was set forth in 1690 by Dutch physicist Christiaan Huygens. Every point on a wavefront serves as a source of spherical, secondary wavelets; the wave at a later time is just the sum of these secondary wavelets.

Figure 2. Secondary wavelets add up to recreate the primary wavefront, later in time.
Gustav Kirchoff showed in the nineteenth century that Huygens' principle can be derived from the wave equation, which in turn is a consequence of Maxwell's equations, finally putting the concept on a firm mathematical foundation.
Far from the source of a spherical wavelet, the wavelet looks like a plane wave; plane waves are easily represented in complex notation:
![]()
When light passes through a grating, i.e. an object or screen that is opaque in some places, we can use Huygen's principle to determine what the wave disturbance looks like far away from the grating. If the grating is two-dimensional, the wave is simply the sum of all the Huygen's wavelets emanating from the transparent parts.
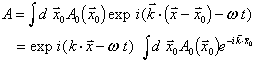
(You may notice that this is just the Fourier transform of the distribution of points that can serve as wavelet emitters.)
Consider a grating of alternating clear and opaque bars. To simplify the calculation, we can treat each clear bar as the source of one Huygens' (now cylindrical) wavelet. This problem then reduces to the multiple slit diffraction problem, and the sum of the wavelets will vanish unless the path length difference from successive bars is a multiple of the wavelength. There will several diffraction orders corresponding to differences of zero, one, two, three…wavelengths.
Laboratory Task 4
Use the HeNe laser (l = 633 nm) to estimate the spacing of the smallest and the second smallest pattern on the microscope resolution test slide. (Use care _ the resolution test slide is quite expensive.) The microscope objective captures the light transmitted by the grating and refocuses it, putatively making an image of the grating. But the microscope objective may not be large enough to capture all of the diffraction orders. In a worst case scenario, no non-zero orders are captured by the objective. What would the image look like if only the zero order diffracted beam is used to form an image? What would the image look like if only the zero and first order beams are used?
Laboratory Task 5
Using multiple slit diffraction theory, calculate the size an objective lens needs to be to capture at least the first order diffraction beam from a grating. The grating has a periodicity of L, and the objective is located 1 mm from the grating.
It should be clear that the resolution of a microscope (i.e. the ability to make an image of a narrowly spaced grating) depends on both the wavelength of the illuminating light and on the "size" of the objective lens. To make any intensity variation at all, the objective needs to capture at least the first order diffraction line. Since this line emanates from the grating at a particular angle, it is the angle that the objective subtends (as viewed from the sample) that is important. The numerical aperture of an objective is defined as n sin q , where q is the angle to the edge of the objective, measured from the optical axis, and n is the index of refraction of the intervening medium, usually air, but sometimes oil (n=1.5). Resolution is formally defined as
d = 0.61 l / N.A.
Laboratory Task 6
Estimate the numerical aperture of the 10X objective. Question: Why does the N.A., and consequently the resolution, depend on the index of refraction of the intervening medium?
If all the diffraction orders are captured and used to form an image of a grating, then the image will generally be quite sharp (but not infinitely sharp!) If the grating is very small, there will only be a few diffraction orders. For example, if the bright zones are less than two wavelengths apart, then there will only be three diffracted beams, the zero order and the two first-order beams. In this case, the image will have a sinusoidal variation in intensity, not a "square wave" variation.
Thought question (to discuss in your reports): why can we simplify our analysis of a microscope to its behavior with simple gratings?
Phase Contrast
If a grating is made of materials that are not opaque but that differ in refractive index, then there will still be diffracted beams, but they will be recombined by the objective to give an invisible image, which is useless. Zernike's insight was that phase differences could be converted into intensity differences if the undiffracted light were phase-shifted. The best way to visualize this is with a phasor diagram. Any beam of light has a phase and an amplitude, so it can be graphically represented as a "vector", where the length is proportional to the amplitude and the angle from the x-axis is the phase. If two beams of light combine, we simply use vector addition on these phasors to determine the final amplitude and phase. If a transparent phase grating is placed in the microscope, then the diffracted beams, plus the zero-order (undiffracted) beam, add to a uniform intensity across the image. It turns out that the higher order diffracted beams are 90° out of phase with the zero-order beam, and are considerably weaker, leading to the phasor diagram shown below. If the zero-order beam can be attenuated and phase shifted, then the changes in the higher-order beams will appear as intensity changes, i.e. as changes in the overall length of the phasor sum. This is the role of the phase ring in the condenser and the phase plate in the back focal plane of the objective. After aligning the condenser for Kóhler illumination, the phase ring (Ph 1 for the 10X objective) is aligned to the phase plate in the objective using the telescoping eyepiece.
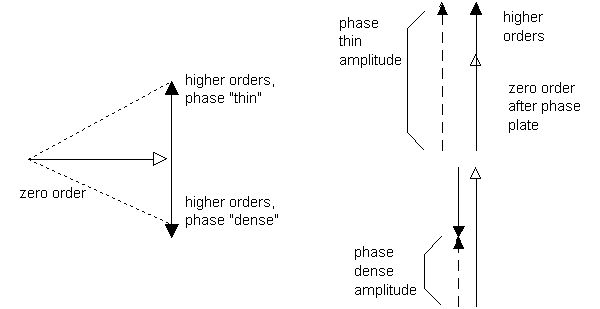
Figure 3. Phasor diagram of light from a phase grating, left, and after passage through the phase plate, right.
Laboratory Task 7
Align the optics for phase contrast. Observe the specimen of fixed cells in both brightfield and phase contrast optics. Describe the differences in the images.
Laboratory Task 8
(May be done at home after class.) Draw a ray diagram of the microscope, properly aligned, in bright field microscopy. Use dotted lines to show rays emanating from the filament, and solid lines to show rays emanating from the sample.
Group laboratory reports
Group laboratory reports are due in one week after completing the lab. Discuss the tasks you performed, using quantitative analysis where appropriate. Be sure to discuss all questions asked in this handout.