Abstract
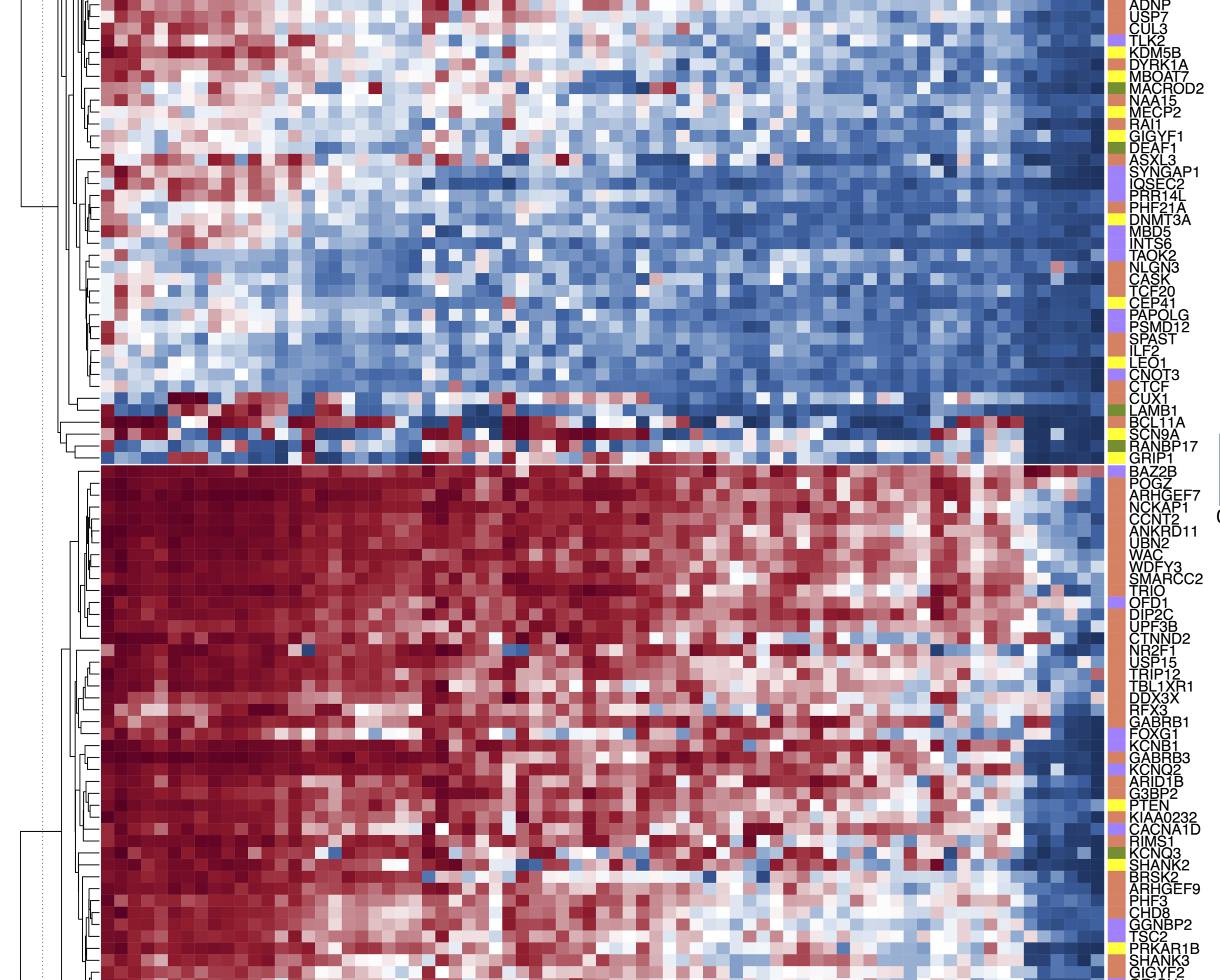
Autism spectrum disorder (autism) is a condition with strong but heterogenous genetic contribution. Recent exome and genome sequencing studies have uncovered many new risk genes through de novo variants. However, a large fraction of enrichment of de novo variants observed in cases are not accounted for by known or candidate risk genes, suggesting that the majority of risk genes are still unknown. Here we hypothesize that autism risk genes share a few common cell-type specific gene expression patterns during brain development, and such information can be quantified to improve statistical power of detecting new risk genes. We obtained large-scale single-cell RNA-seq data from human fetal brain collected through a range of developmental stages, and developed a supervised machine-learning approach "A-risk" (Autism risk), to predict the plausibility of autism risk genes across the genome. Using data from recent exome sequencing studies of autism, A-risk achieves better performance in prioritizing de novo variants than other methods, especially for genes that are less intolerant of loss of function variants. We stratified genes based on A-risk and mutation intolerance metrics to improve estimation of priors in extTADA and identified 71 candidate risk genes. In particular, CLCN4, PRKAR1B, and NR2F1 are potentially new risk genes with further support from neurodevelopmental disorders. Expression patterns of both known and candidate risk genes reveals the important role of deep-layer excitatory neurons from adult human cortex in autism etiology. With the unprecedented revolution of single-cell transcriptomics and expanding autism cohorts with exome or genome sequencing, our method will facilitate systematic discovery of novel risk genes and understanding of biological pathogenesis in autism.Competing Interest StatementThe authors have declared no competing interest.