Abstract
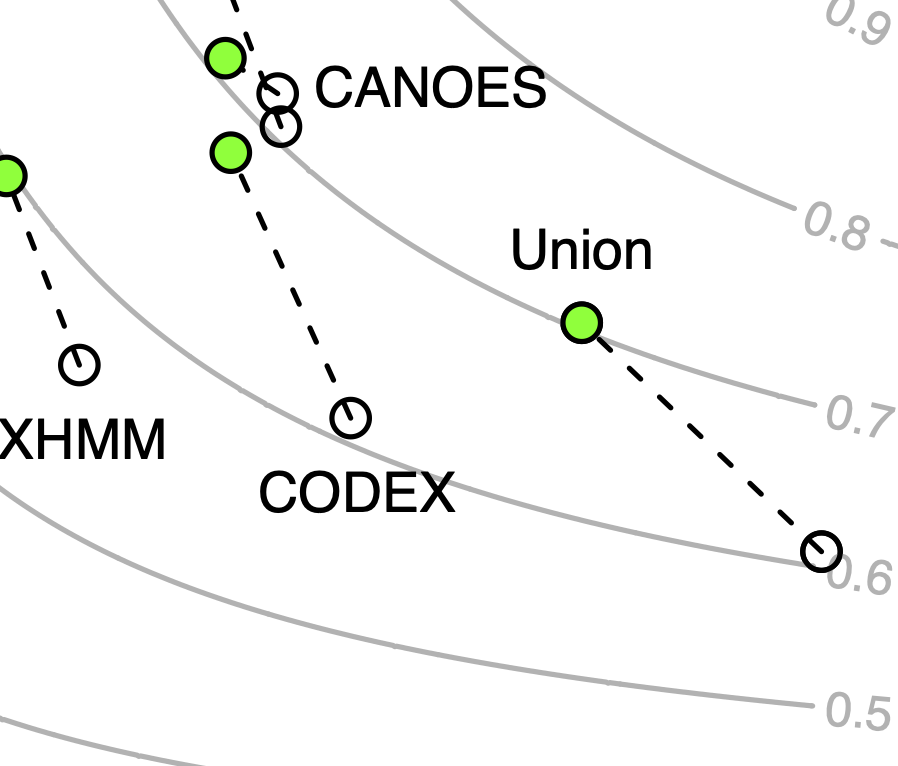
Exome sequencing has been widely used in genetic studies of human diseases and clinical genetic diagnosis. Accurate detection of copy number variants (CNVs) is important to fully utilize exome sequencing data. However, due to the nature of noisy data, none of the existing methods can achieve high precision and high recall rate at the same time. A common practice is to perform filtration with quality metrics followed by manual inspection of read depth of candidate CNV regions. This approach does not scale in large studies. To address this issue, we present a deep transfer learning method, CNV-espresso, for confirming rare CNVs from exome sequencing data in silico. CNV-espresso encodes candidate CNV regions from exome sequencing data as images and uses convolutional neural networks to classify the image into different copy numbers. We trained and evaluated CNV-espresso on a large-scale offspring-parents trio exome sequencing dataset, using inherited CNVs in probands as positives and CNVs with mendelian errors as negatives. We further tested the performance using samples that have both exome and whole genome sequencing (WGS) data. Assuming the CNVs detected from WGS data as proxy of ground truth, CNV-espresso significantly improves precision while keeping recall almost intact, especially for CNVs that span small number of exons in exome data. We conclude that CNV-espresso is an effective method to replace most of manual inspection of CNVs in large-scale exome sequencing studies.
A preprint version is available on bioRxiv: https://doi.org/10.1101/2022.03.09.483665