Stretching Bonds
Bond lengths between pairs of atoms in covalent molecules are generally predicted well by the sum of their respective covalent radii, such that there are usually only small variations in related compounds. It is, therefore, significant that we have demonstrated that the incorporation of appropriately sized linkers between carbon and a metal center provides a means to modulate the length and nature of a metal–carbon interaction.
Specifically, whereas [Tptm]ZnX compounds have similar Zn–C bond lengths (2.11 – 2.19 Å), the Zn–C bonds in [TitmMe]ZnX compounds are much longer and highly variable (2.44 – 2.68 Å).
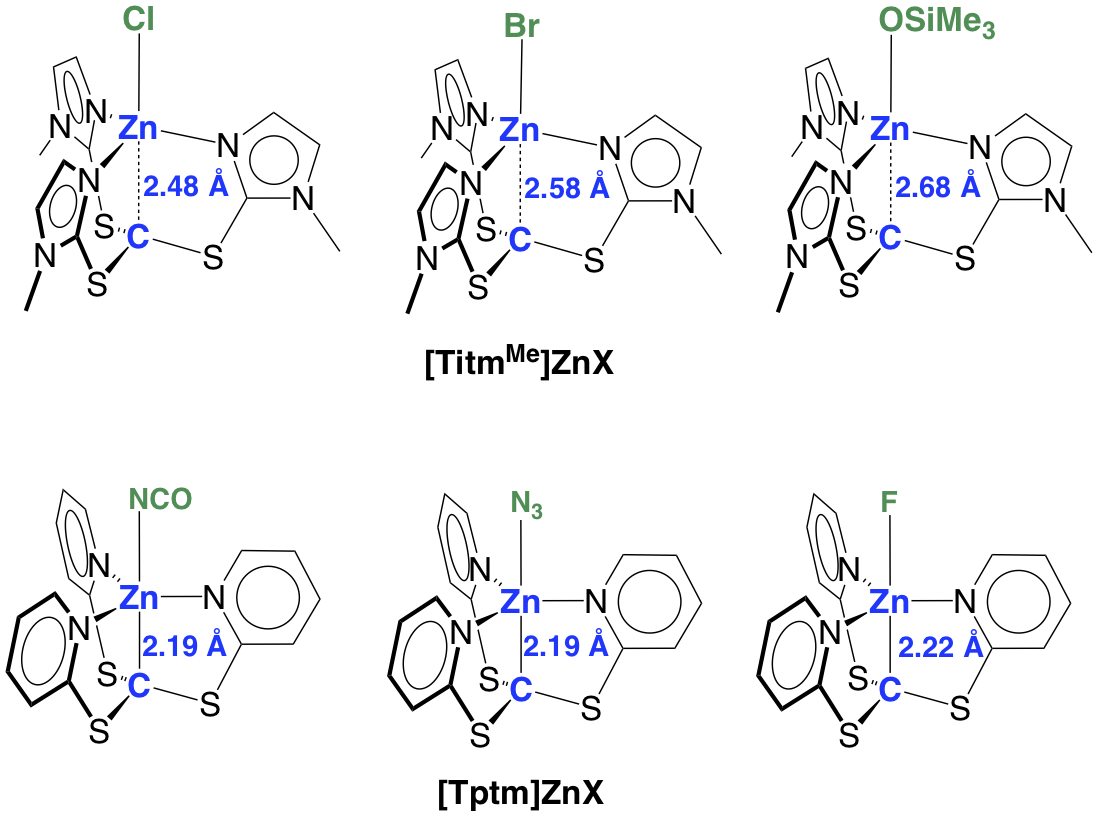
The unusually long Zn–C interactions in [TitmMe]ZnX cannot be described to conventional covalent bonds and are proposed to be indicative of a significant ionic component, such that the compounds are best described as zwitterionic, [C– Zn+–Cl], with formally anionic and cationic carbon and zinc centers, respectively. Density functional theory (DFT) calculations indicate that the HOMO of [TitmMe]ZnCl is effectively an spn orbital localized on carbon, which is in accord with the zwitterionic description.
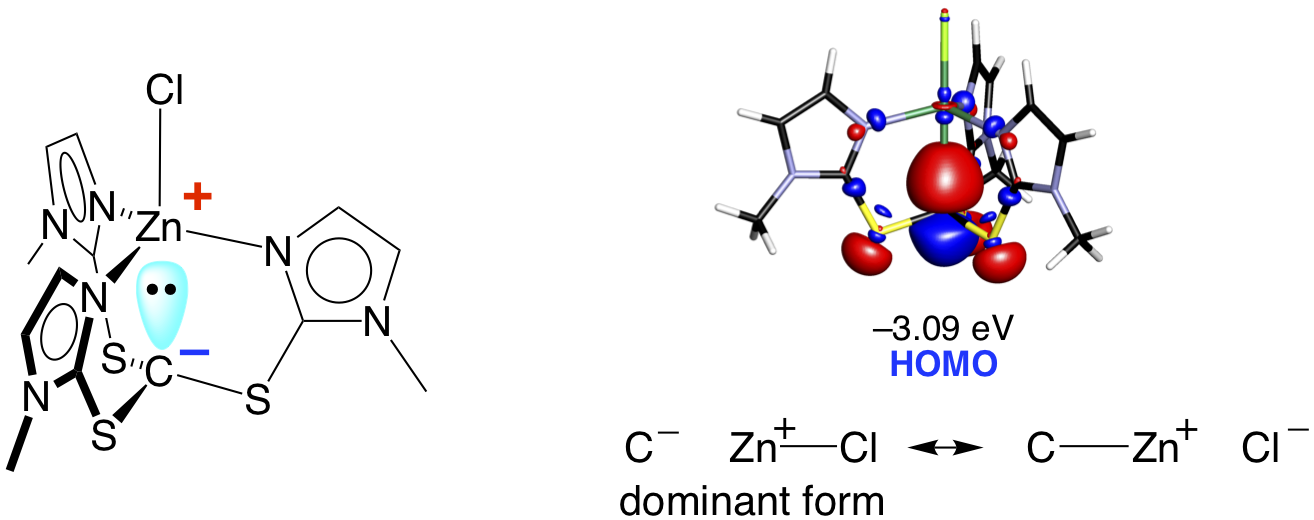
Furthermore, DFT studies demonstrate that the energy surface associated with varying the Zn–C bond length of [TitmMe]ZnCl is rather shallow, thereby rationalizing the variable Zn–X distances.

The origin of the electronic difference between [TitmMe]ZnCl and [Tptm]ZnCl is attributed to the different angular requirements of the 5- and 6-membered heterocyclic rings, which cause the zinc to be displaced from the plane of the three nitrogen atoms in the former compound, thereby lengthening the Zn–C bond. The ability to modify the electronic nature of an M–C bonding interaction in this way is a novel aspect of carbatrane chemistry.

• Ruccolo, S.; Sattler, W.; Rong, Y.; Parkin, G. “Modulation of Zn–C bond lengths induced by ligand architecture in zinc carbatrane compounds” J. Am. Chem. Soc. 2016, 138, 14542−14545.