(With embedded Q&A)
Lec. 4. Biol C2005/F2401 L. Chasin
Sept. 16, 2010
Last updated:
Thursday, September 16, 2010 12:21 AM
© Copyright 2010 Lawrence Chasin and Deborah Mowshowitz
Department of Biological Sciences Columbia University
New York, NY
Handout:
None (have #3
already)
Props: 2 aa
models, 3 ropes ...
Lipids, cont:
Fats
Phospholipids
phosphoester bonds
lipid bilayer
biological membranes
Proteins: monomers = amino acids
pK
Stereoisomers: L and D amino acids
Protein structure (4
levels):
Primary (1o)= linear sequences of AAs
Polypeptides, peptide bond, backbone, N-terminal, C-terminal
Methods:
Paper electrophoresis of amino acids
Paper chromatography of amino acids
Fingerprinting: 2-dimensional electrophoresis + chromatography of small peptides
Sickle cell disease: an altered sub-peptide of the globin polypeptide
Primary (1o) structure = AA seq
3D structure is reproducibly formed.
Denaturation, renaturation
Fats: Fatty acids and
glycerol as monomers)
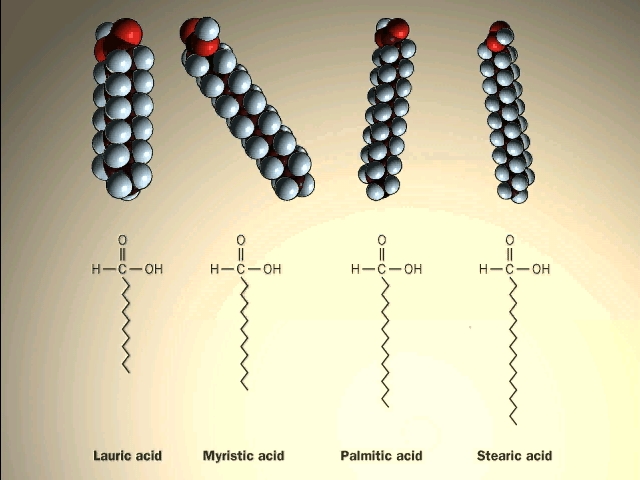
A major class of lipids are the fatty
acids, long straight chain hydrocarbons with a carboxyl group (carboxylic acid)
on one end. See
[Purves 6:3.19, 7:3.18], and another
picture.
{Q&A}.
![]()
(Esters; fats vs.
oils; saturated vs. unsaturated fats)
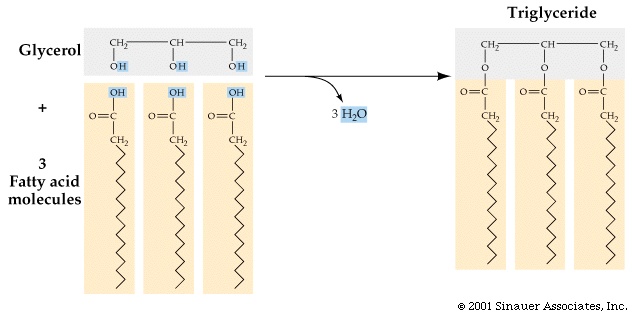
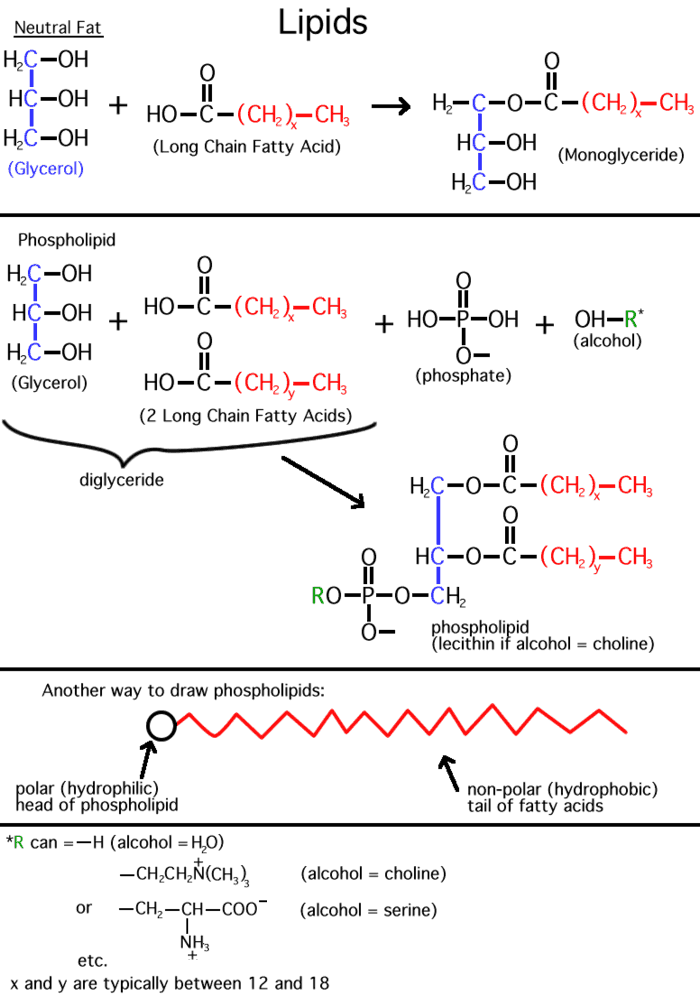
Inside cells, fatty acids (FA) are usually connected to a molecule of the
tri-hydroxy (tri-alcohol) compound glycerol. Once again water is removed, this
time producing an ester bond (acid
+ alcohol, see top right corner of
lipids handout 2-10). If all 3 OH 's on the glycerol are
substituted with FA's, then we have a triglyceride. See
[Purves 6:3.19, 7: 3.18], and another
picture. This is fat. Fats are very hydrophobic and
are practically insoluble in water. You can also have mono- or di-substituted
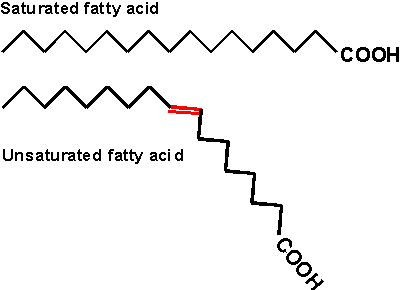
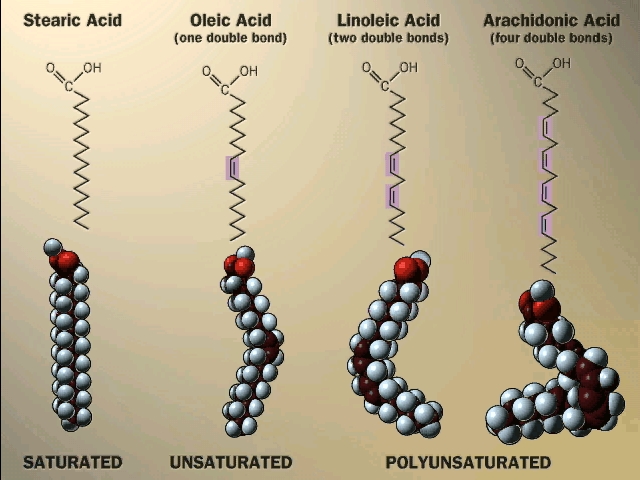
glycerol, but it is the triglyceride that is fat. Fats differ according to the exact nature of the FA's that are
present. "Saturated" fats have -CH2- (methylene) groups, usually
18-20 of them, along the chain. They are saturated with hydrogens,
compared to the unsaturated variety. The latter may have a double bond or two within
the chain, and thus have less H's (unsaturated for H's). The presence
of the double bond puts a crimp into the structure, since unlike single C-C bonds,
there is no rotation about C=C double bonds)
[Purves 6:3.20, 7:3.19]. As a result, it is more difficult for the unsaturated fatty acid molecules to
associate.
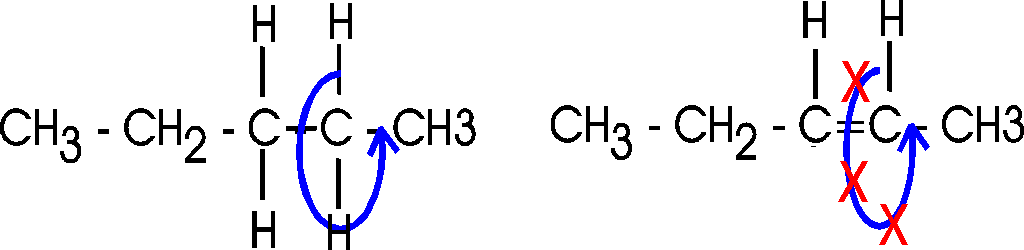
Actually, there are 2 ways a double bond can form in a fatty acid, called cis and trans. In the cis case, the two hydrogens are on the same side of the double bond (remember there is no rotation, so their position is fixed). Now the two bonds carrying the rest of the carbon atoms are also together on one side of the double bond, so the molecule is crimped, with a severe angle between the two hydrocarbon stretches. In the trans case, the two hydrocarbon stretches are on opposite sides of the double bond, and the overall chain is straighter. Most fatty acids in animals are saturated; with their relatively straight chains their hydrocarbon chains are free to associate with each other with no constraints and they aggregate into solid fat. Plants contain a lot of unsaturated fatty acids of the cis type. These unsaturated fats (WITH the double bonds) are usually liquids (oils), as their crooked fatty acid chains cannot approach each other so easily. Take vegetable oil (unsaturated), and add hydrogen across the double bonds and you get Crisco, or the creamy texture in peanut butter (read the label: hydrogenated).
![]()
Trans fatty acids do occur made by bacteria in the stomachs of ruminants like cattle, so they end up in beef to some extent. A much greater source of trans unsaturated fatty acids comes from the chemical hydrogenation of oils, where they are formed somewhat ironically as a by-product of the hydrogenation process. The trans unsaturated fats resist turning rancid, so are favored by the food industry. However, they are more equivalent to saturated fatty acids in their ability to form solid fat, which encourages the formation of atherosclerotic plaques. Thus margarine may be as bad for you as butter. Or worse, as trans fats show a stronger correlation with heart disease than saturated fat, possibly through more indirect effects (e.g., membrane structure).
So here again as in the case of polysaccharides, the 3-dimensional structure of the molecule has a lot to do with its physical properties.
Fats are a good example of hydrophobic forces at work. Just think of a fatty chicken soup with those globules of fat floating on top, out of solution.
Fats serve as a storage form of energy. That is, like glycogen or starch, fats can be broken down and used for energy metabolism, as we will see later. Fats are stored in cells called adipocytes.
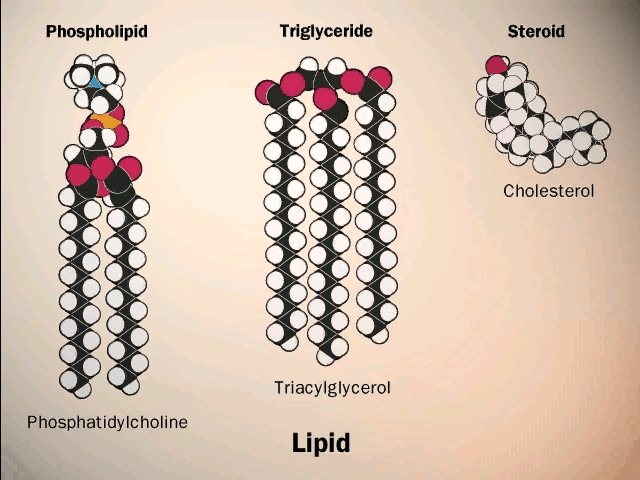
(Phospholipids,
phosphoesters, phosphatidylcholine)
There is a special class of lipids that are
related to the fats, but with a significant difference. These are the
phospholipids, an
example of which is shown in the middle of the
LIPIDS handout. Two of the glycerol hydroxyls are connected to long chain
fatty acids, but the third is connected to quite a different group, a phosphate.
Phosphoric acid (H3PO4) is an acid, The -OH groups
attached to the phosphorous easily lose hydrogens at neutral pH.
Phosphoric acid has 3
acidic hydrogens. [If you are shaky on pH, ask to review it in recitation
section.]
Phosphoric acid is a strong acid, losing most of its hydrogen ions at pH7. The ion that is formed is called phosphate, and we will treat the 2 names equivalently, considering them both acids (referring to their origin as the acid). Similarly we will use carboxylic acid and the carboxylate ion (the negatively charged unprotonated form) synonymously in most situations.
The phosphate group is connected to a glycerol hydroxyl, again by a dehydration that forms an ester (acid + alcohol). Whereas up until now we had a carboxylic acid ester linking the fatty acid to the glycerol, here we have a phospho-ester. The acid partner is the one named to specify a type of ester. In both cases the ester is formed by an alcohol linked to an acid. After linkage, the phosphate group is still charged, as shown. The rest of the phosphate may be free, as in a phosphatidic acid, or it may be esterified to yet another alcohol via another of its acidic groups; a common one is ethanolamine: HO-CH2-CH2-NH3+. The resulting phospholipid would be called phosphatidyl-ethanolamine, and it would be categorized as a phospho-di-ester (phosphodiester). Note that the presence of positively charged basic groups such as amines tends to neutralize the negative charge of the phosphate, but only adds to the hydrophilic character of the head of the phospholipid, by adding charged groups.
Phospholipids,
phosphoesters, phosphatidylcholine)
There is a special class of lipids that are
related to the fats, but with a significant difference. These are the
phospholipids, an
example of which is shown in the middle of the
LIPIDS handout. Two of the glycerol hydroxyls are connected to long chain
fatty acids, but the third is connected to quite a different group, a phosphate.
Phosphoric acid (H3PO4) is an acid, The -OH groups
attached to the phosphorous easily lose hydrogens at neutral pH.
Phosphoric acid has 3
acidic hydrogens. [If you are shaky on pH, ask to review it in recitation
section.]
Phosphoric acid is a strong acid, losing most of its hydrogen ions at pH7. The ion that is formed is called phosphate, and we will treat the 2 names equivalently, considering them both acids (referring to their origin as the acid). Similarly we will use carboxylic acid and the carboxylate ion (the negatively charged unprotonated form) synonymously in most situations.
The phosphate group is connected to a glycerol hydroxyl, again by a dehydration that forms an ester (acid + alcohol). Whereas up until now we had a carboxylic acid ester linking the fatty acid to the glycerol, here we have a phospho-ester. The acid partner is the one named to specify a type of ester. In both cases the ester is formed by an alcohol linked to an acid. After linkage, the phosphate group is still charged, as shown. The rest of the phosphate may be free, as in a phosphatidic acid, or it may be esterified to yet another alcohol via another of its acidic groups; a common one is ethanolamine: HO-CH2-CH2-NH3+. The resulting phospholipid would be called phosphatidyl-ethanolamine, and it would be categorized as a phospho-di-ester (phosphodiester). Note that the presence of positively charged basic groups such as amines tends to neutralize the negative charge of the phosphate, but only adds to the hydrophilic character of the head of the phospholipid, by adding charged groups.
(Phospholipid bilayer:
membranes)
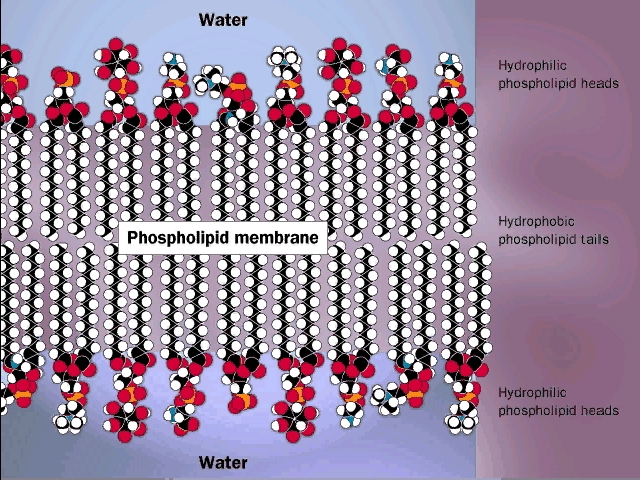
Phosphatidyl-ethanolamine is a compound
that is highly hydrophobic throughout most of the molecule, but then has a
highly polar group at one end, with two complete, if opposite, charges. A
further derivative has 3 methyl (-CH3) groups bonded to the
nitrogen instead of H's. This moiety is choline (tri-methyl-ethanolamine; the
nitrogen retains its positive charge. When esterified to a diglyceride one gets
phosphatidyl choline, depicted in [Purves
6:3.21, 7:3.20]. The polar end can interact strongly with water (it is hydrophilic),
while the remainder of the molecule wants to come out of aqueous solution. This
is a confused molecule. What happens is that the hydrophobic parts all line up
with each other to minimize their interface with water (both side-to-side and
end-to-end), while the charged ends remain in contact with water. See
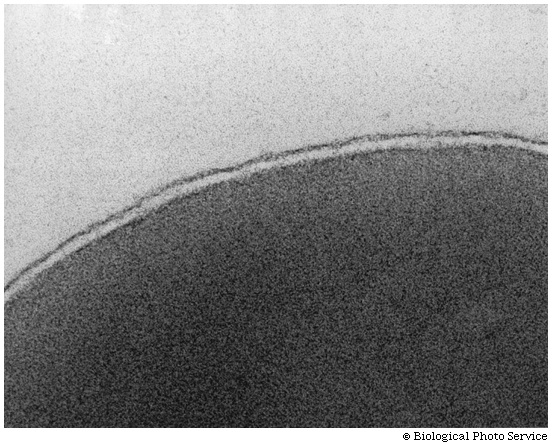
[Purves 6:3.22, 7:3.21], and
photo. It is in this way that biological membranes form, as
a phospholipid bilayer, the charged ends of the double layer
being on the outside in contact with water, with the cytoplasm on one side and
the exterior of the cell on the other side: See
picture. And look in your textbooks for great diagrams of phospholipid
bilayers.

Such a bilayer presents a permeability barrier to water-soluble compounds, which cannot pass through the hydrophobic barrier. Special protein structures that are embedded in this membrane are then necessary to allow the passage of water soluble compounds in and out of the cell. These are the channels and pumps mentioned earlier. See a diagram of a cell membrane at [Purves 6:5.1, 7:5.1] and in your texts. Yet again we see how the chemical properties of these molecules determine their structure, and how their structure provides a biological function. To review phospholipid structure, try problems 1-12 & 1-13.
Large amounts of cholesterol are embedded in the membranes of animal cells. The cholesterol is kept inside by hydrophobic forces. It acts to plug spaces that could cause leakiness, to impart more strength, and to prevent too much association of the saturated fatty acids at low temperature (i.e., "freezing" of the membrane into fat).
The texts have nice diagrams of all this.
Lipids are impressive in their variety (see picture) and especially in membrane formation, but admittedly they are not really good examples of the linear biopolymers that we defined. But they have to go somewhere, and so they are stuck amongst the macromolecules.
NUCLEIC ACIDS: Unlike the catch-all category of LIPIDS, NUCLEIC ACIDS are biopolymers par excellence. There are 2 types, DNA and RNA, the monomers are nucleotides, that have nitrogen-containing rings, 5-carbon sugars, and phosphodiester linkages. There are four types of monomers in each polymer. We will discuss them in detail, but not for a few weeks yet.
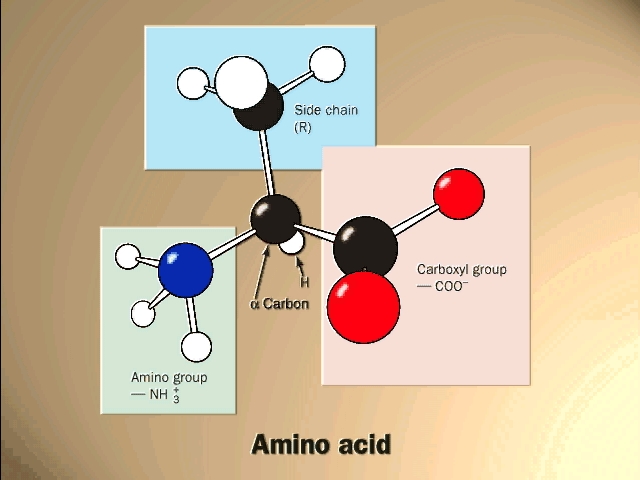
(Proteins: Amino acids are the monomers (20))The general structure of an amino acid is:
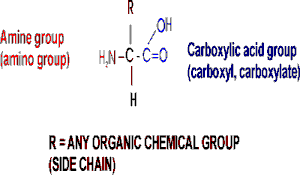
Note the central carbon atom, to which 4 different groups are attached: an amino group (drawn by convention at the left), a carboxylic acid group (put at the right side), a hydrogen, and a side chain, or R-group. Only the R-group varies among the 20 different amino acids. This is the side chain, and so there are 20 different side chains. Look at the amino acids and peptides handout for some of the side chains. Your texts and hard copy handout show all 20, and you should examine all 20.
Out of laziness, I drew the general amino acid incorrectly: Actually at neutral pH, the molecule is charged, because the carboxylic acid group is an acid, and the amine group is a base, so more accurately: (also see 3-D picture)
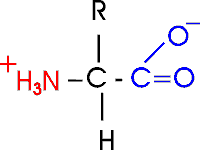
Let's take this opportunity to discuss the charge on
organic molecules a bit more. In living systems, the carboxylic acid group
is mostly charged and the amine is mostly charged, but that is at pH7, the
cellular pH under most circumstances. Is an acid always mostly charged in aqueous
solution? No. It depends on the pH of the environment. In the laboratory we do
not have to keep things at pH 7, as it is in the cell. We can vary the environment at will, adding strong acids such as hydrochloric acid as a source
of hydrogen ions (lowering the pH), or a strong base such as sodium hydroxide
(raising the pH). The strength of an acid is a measure of how readily it gives
up a proton. Carboxylic acids are always in equilibrium with the hydrogen ions
(protons) in the solution, so if the hydrogen ion concentration is high (acidic)
then the equilibrium will shift toward the protonated (uncharged) species. At pH
2.5 an amino acid carboxyl group is protonated about half the time; for each pH
unit this proportion of protonated species will drop by a factor of 10, so very
little of the carboxyl group is protonated at the neutral pH of 7 found in most
cells. A similar situation pertains to the amine base end: at a very low H+ ion
concentration (e.g., 10-11 M H+, a high pH of 11), it will tend to
lose its extra proton, but at pH 7
it will mostly remain protonated, with a positive charge. There will always
be some intermediate pH at which we find the the group half charged/half
uncharged. This pH is called the pK of the group, and it can be influenced
by the remainder of the molecule. The pK is an indication of the acidic or basic
strength of the group (the lower the pK the stronger the acid, the higher the pK the stronger the base).
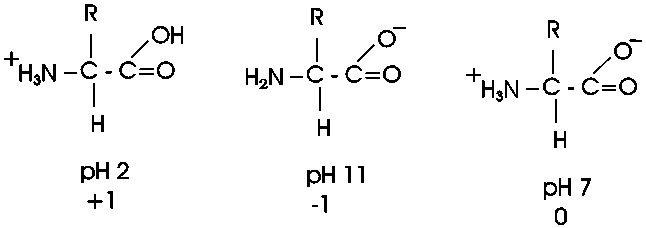
So at pH7, most amino acids are neutral (no net charge), but they are highly charged nonetheless. {Q&A}
A molecule that is charged but electrically neutral is called a zwitterion.
Now, what are some of these 20 different side groups?
Here are 2 charged side group, e.g.:
asp: R= -CH2-COO- , there is a
second carboxyl group on this amino acid)
lys: R= -CH2-CH2-CH2-CH2-NH3+
, there's a second amine on lysine, so lysine will have 3 charged groups,
and a net charge of +1 (two +'s and one -) at pH7.
There is a convention for numbering amino acid carbons; actually it's a lettering. It starts from the central carbon, called alpha: so lys has (count with me) an alpha, beta, gamma, delta, EPSILON-amino group as well as an alpha-amino group (and an alpha-carboxyl).
The average molecular weight of an amino acid is ~120, but the range is from 75 to 203.
The smallest amino acid (a.a.) is glycine (gly), MW = 75. Here the side chain is merely hydrogen.
The largest is tryptophan (trp), MW = 203 [-CH2- bridge to a 5-membered ring containing a N plus a fused 6-membered ring] and is fairly hydrophobic.
Two of the amino acids contain sulfur, the first instance we have seen of that element.
Note the difference between the acidic side chains of aspartic acid and glutamic acid and the amide versions of these amino acid, asparagine and glutamine. The latter two ate very poalr but not charged.
Look over the structures of the 20 amino acids in the textbook. It is the properties of the functional groups on the 20 different side chains of the 20 different amino acids that determine the function of a protein, so they are all-important. The handout shows all 20 aa's, but without indicating the ionization of the acidic and basic groups. We will discuss many of the side chains within the context of the discussion as we go along.
There are two that deserve special mention: arginine contains a functional group that will not be found elsewhere in this course; it is -NH-C+(NH2)NH2, called the guanido group. The guanido group is a strong base, even stronger than an ordinary amine, so it is positively charged at pH7 (like lysine). You can consider the positive charge to be distributed over all 3 N atoms. Proline has a side chain that folds back and forms a covalent bond to the amine nitrogen of the amino acid, thus producing a ring structure.
You should be able to recognize the properties of the side chains as polar or non-polar, charged or not charged. You will not be responsible for recalling a specific amino acid structure from the English name or vice versa, but given the structure you should know how it behaves. {Q&A}. Although it is not required in this course, it would be a good idea to memorize the 3-letter and 1-letter abbreviations of the amino acids, as it makes communication and reading easier. Especially if if you plan to take upper level biology courses.
To be sure you understand amino acid structure, try problem 1-15 except C. For additional review of the effects of pH, try problem 1-16. For the effects of different R groups, try 1-20.
(Stereoisomers)
Now let's consider the structure of an amino acid in
3 dimensions:
When carbon forms 4 single bonds, it makes them spaced equally apart from each other in space, in the form of a tetrahedron as in this representation of alanine.
Now consider this other molecule of an amino acid [again with 2 white groups], with 2 H's of glycine, e.g. Are these the same molecule, that is, are they distinguishable or are they indistinguishable?
They are indistinguishable, since I can rotate them and superimpose their atoms.
But now suppose I switch the positions of two of the atoms attached to the central carbon. Now I can no longer superimpose them, so they are two distinct molecules, different from each other in 3-dimensonal space. Surely their chemical properties should be virtually identical since each has the very same 4 groups attached to a central carbon. They are both alanine, as they have the same four groups attached to the central carbon. But in three dimensions they are actually mirror images of each other. See [Purves6ed 2.21a]. We call one D-ala and one L-ala. See [Purves6ed 2.21b].
This one is D, or is it this one... ? I can't remember .. it's not too important here.
What is important is that in general, you have this situation, the possibility of two stereoisomers, whenever there is what is called an ASYMMETRIC carbon atom in a molecule, that is, a carbon with four different groups attached.
These stereoisomers are sometimes called optical isomers, since the two forms, in solution, will bend a beam of polarized light one way or the other. Thus the D designation originally meant dextro, or to the right, whereas L stood for levo, to the left.
All amino acids except glycine have an asymmetric carbon, which is the alpha-carbon. So we can draw 19 of the amino acids in 2 stereoisomeric forms.
So do we really have 39 a.a.'s? No. All the stereoisomeric forms of the amino acids in proteins are L-amino acids, so we only have to worry about 20. D- amino acids do occur occasionally in nature, but not in proteins
Note that the sugars we discussed, like glucose, have several asymmetric carbon atoms. Aside from L and D designations, the sugar stereoisomers are given different English names (e.g., D-glucose, D-mannose, L-rhamnose, etc.).
To review the various chemical groups discussed so far, try problem 1-19.
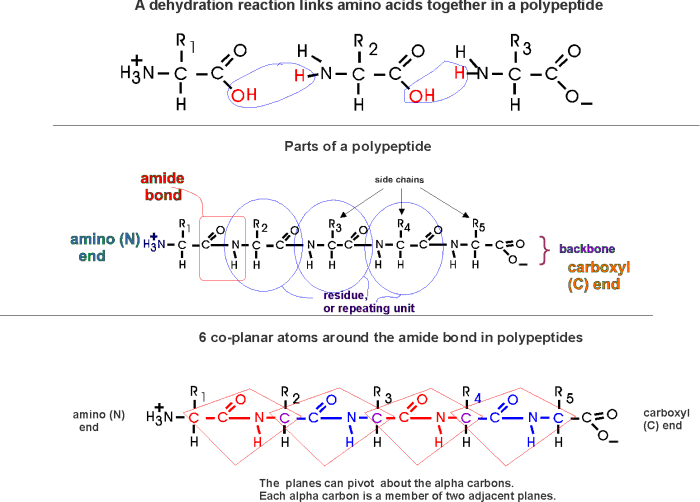
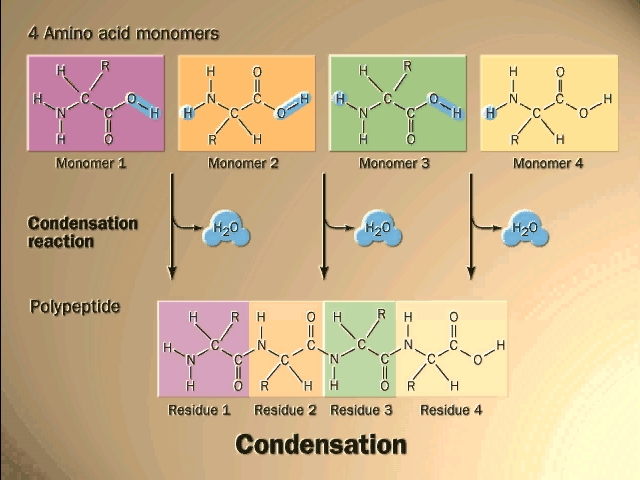
(Polypeptides, peptide bond)
Polymerization of aa's
OK, now let's now string these L-amino acids together, polymerize them. The
bond that connects two amino acids is an AMIDE bond
(-CO-NH-) between the carboxyl of one amino acid and the amino group of the
next. Once again, a molecule of water is removed in the formation of the
connecting bond:
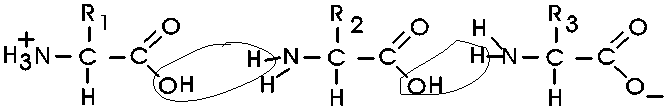
In the special case of proteins, this amide bond is called a PEPTIDE BOND, and the resulting product a PEPTIDE, a dipeptide (or we could go on to a tri-peptide, oligo-peptide, or finally, POLY-PEPTIDE). (See also polypeptide handout). Also see [Purves6ed 3.4], and another picture. {Q&A}
By convention, the amino group is written on the left for an amino acid and also for a peptide.
In the tripeptide in the diagram, note the peptide bond (boxed), and the repeating unit, or aa "residue" [circled]. Residue refers to what's left of the amino acid monomer after it has been incorporated into a polypeptide, which is most of it: it just lacks one H at what was the amino end and one OH at what used to be the carboxyl end. Note also that the charged amine and carboxyl groups no longer exist inside the polypeptide, having been replaced by the amide, an uncharged (but polar) functional group.
Almost all polypeptides have 2 ends, the amino end and the carboxyl end, which do remain charged at pH7.
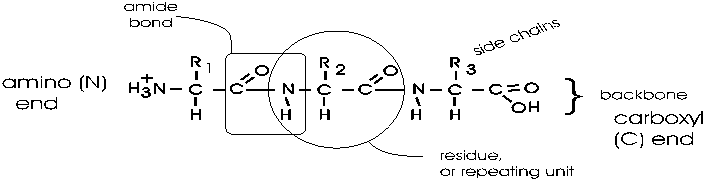
The "backbone" of the polypeptide is defined as all of the atoms except the side chains.
The only free amino and carboxyl atoms of the backbone are at the 2 ends.
The side chains , then, stick out of this backbone (also see polypeptide handout).
Nomenclature: e.g., alanine-methionine-alanine, or ala-met-ala, or alanyl-methionyl-alanine, or AMA
To review peptide structure, try problem 1-15, C, and then try 2-3 part A.
The length of polypeptides is commonly 100-1000 amino acids, but smaller and larger ones also can be found.
Each and every protein molecule in the cell has an identity defined by its particular sequence of amino acids. Each E. coli cell contains about 3 million polypeptides molecules, but only about 3000 different ones. Each of these individual protein types has a name to go along with its chemical identity.
Are there enough combinations to specify 3000 different polypeptide sequences? Well, if the average polypeptide is 500 amino acids long, then the possible combinations are 20500 or 10650, which is a number of inconceivable magnitude. So evolution has settled on about 100,000 of these combinations to do biological jobs.
Some examples of polypeptides, taken not from E. coli, but from more familiar organisms include:
Carriers: hemoglobin, which carries oxygen in red blood cells;
Nutrients: egg albumin, a nutrient in the white of a hen's egg;
Structural: keratin, providing toughness in skin, fingernails, and wool;
collagen, providing a strong connection between cells in tendons;
Signal reception: estrogen receptor (intracellular
located)
epidermal growth factor receptor (spanning the cell membrane)
Recognition of foreign substances: immunoglobulins (antibodies)
Enzyme catalysts: beta-galactosidase, which helps digest the milk sugar lactose.
We will discuss enzymes in some detail as an important category of proteins.
(Primary (1o)
structure = linear
sequences of AAs)
Each of these proteins contains a polypeptide with a particular sequence of
amino acids, usually all 20 are represented, although not at all equally. Unlike
polysaccharides, this sequence usually exhibits no obvious simple regularity, or
repeating subsequence:
This linear sequence of amino acids is called the primary (1o) structure of a protein. {Q&A}
It might be, for instance: met-ala-leu-leu-arg-glu-leu-val ...... How is this sequence determined?
(Methods: Paper chromatography,
electrophoresis, fingerprinting)
I will discuss now and next time some
methodology used in the purification of amino acids and proteins. We
bring in some selected lab methods from time to time for two reasons: First, the
behavior of molecules in experimental situations helps you to understand their
behavior in nature; and second, the methodology is interesting in its own right
as an example of how science is done.
Our first topic of methodology is directed at the question of how we get to know this primary structure, this sequence of amino acids in a polypeptide?
The modern way to determine the sequence of a polypeptide involves an instrument called a mass spectrometer: the protein is fragmented and the exact molecular weight of each small fragment is determined by its flight path in an electric field in the instrument. From repetitions of this type of analysis the exact amino acid sequence can be deduced.
A more traditional method used to determine the sequence of a polypeptide is to chemically degrade it from one end in a stepwise fashion, starting at either the amino or the carboxyl end. First you must purify the polypeptide in question away from the other 3000 polypeptides in the cell; we will discuss that process a little later.
The degradation of the polypeptide back to its free monomer AA's is a form of HYDROLYSIS, a reverse of the dehydration that accompanied the formation of the peptide bond. As an example we will only discuss here the degradation of a peptide or polypeptide from the C-terminal. The controlled hydrolysis of amino acid residues from the carboxyl end of a polypeptide is a form of enzymatic hydrolysis; an enzyme, called carboxypeptidase, itself a polypeptide, catalyzes this hydrolysis; it does not happen by itself. Carboxypeptidase is called an exopeptidase since it works on the end (exterior) of the polypeptide. We will learn more about enzymes next week. After the carboxypeptidase is mixed with a peptide, hydrolysis begins: all the trillions of molecules release their C-terminal amino acid in unison, almost synchronously, so that in the first wave the last (original C-terminal) amino acid is released. A bit of the reaction mixture is remove at this point the released amino acid is separated from the main peptide and identified. By letting the reaction proceed for increasing amounts of time, the time that amino acids are released can be correlated with their distance form the C-terminal end.
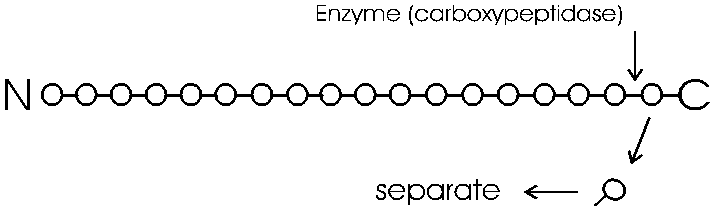
You can get the sequence of perhaps 20 amino acids in from the carboxy terminal in this way, before the process (i.e., the synchrony) breaks down. Since most polypeptides are greater than 20 amino acids in length, you first need to chop the polypeptide into manageable pieces and then sequence each piece by subjecting it to hydrolysis by carboxypeptidase. This internal chopping is done using a different type of proteolytic enzyme, an endopeptidase (cleaves inside, not from the end). One such enzyme is trypsin, which cleaves after the 2 amino acids with basic side groups, arginine and lysine.
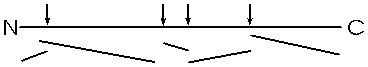
Let's consider the analysis of the sequence of one of
these sub-peptides produced by trypsin cleavage. The problem is how to separate
and identify the different amino acids that are released by this
carboxypeptidase hydrolysis. How do you know which amino acid came off when?
Amino acids behave sufficiently differently from each other under certain
conditions to allow the complete separation of all 20 species from a mixture. We
will discuss two methods for separation and identification here.
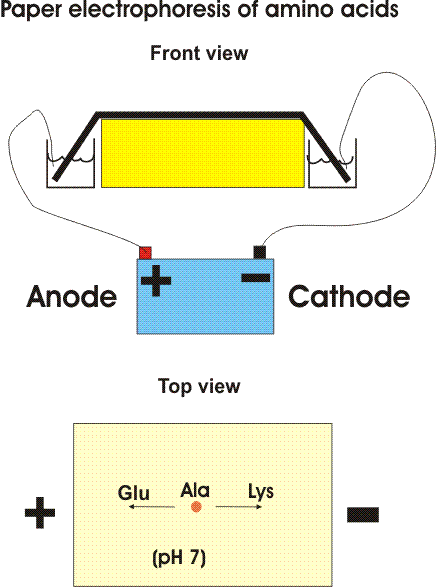
One way is based on the migration of amino acids in an electric field. In PAPER ELECTROPHORESIS, an amino acid mixture is spotted onto a sheet of filter paper, the paper is wet with a buffered salt solution and placed between two electrodes and high voltage (e.g., 2000 volts) applied. At neutral pH, the acidic amino acids (asp and glu) will have a net negative charge and will migrate toward the ANODE (+ pole) while the basic amino acids (arg and lys) will migrate toward the CATHODE (- pole). {Q&A} Electrically neutral amino acids will not migrate much, unless the pH is made acidic or basic (as it is in some problems in the problem book) .
Viewed after application in the center followed by electrophoresis.
Try problem 2-1.
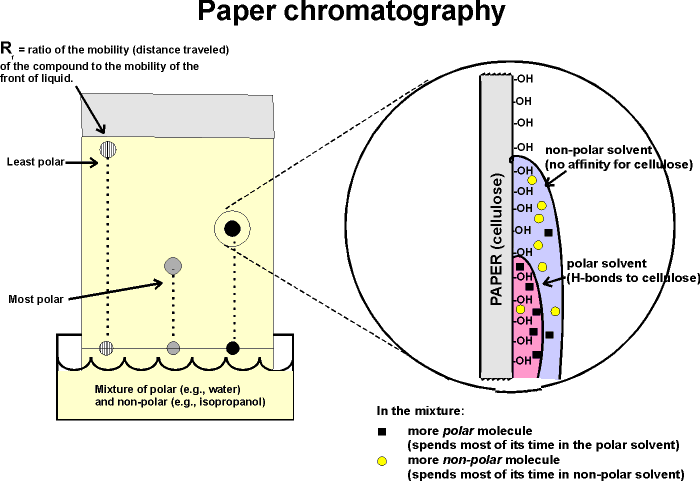
A more versatile separation method is PAPER CHROMATOGRAPHY. This method is based on the differential solubility of the different amino acids
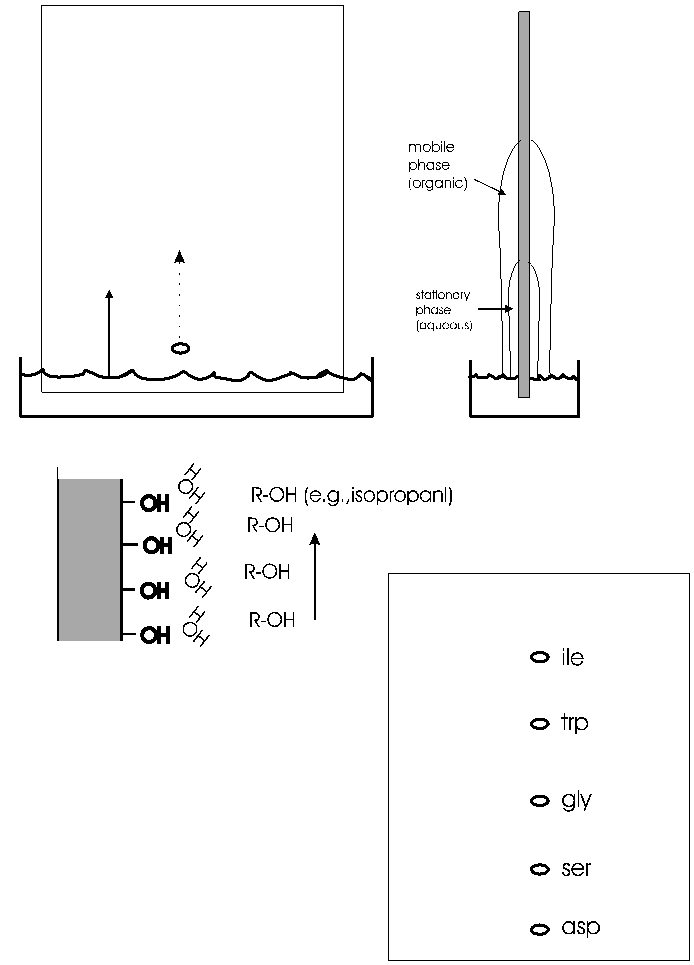
in organic (non-polar) solvents, which in turn is determined by the nature of the side group. The amino acid mixture is spotted onto a filter paper; one edge of the paper is immersed in a mixture of aqueous and non-aqueous solvents. (See handout.) The liquid will be drawn up the paper by capillary action. As it rises, the water in the liquid mixture is bound by the paper (cellulose, with its many OH groups), forming a stationary water layer, or stationary phase. The organic solvent (e.g., propanol) moves up without as much interaction with the solid cellulose; it is considered the mobile phase. The amino acids will be constantly equilibrating between being in the mobile organic phase or the stationary water phase. The more polar the side chain, the more time the amino acid will spend in the stationary phase. The more hydrophobic the side chain, the more time it spend in the mobile organic phase. By using a series of different solvents, all 20 amino acids can be separated in this way. It works for many other organic molecules as well. The distance that an organic molecule moves in a particular chromatographic system is called the Rf, which stands for mobility Relative to the Front, that is, the distance the organic molecule in question migrated divided by the distance that the front of liquid has risen on the paper at that time. Rf's in a particular solvent and at a given temperature are reproducible and are published for many organic compounds, including all the amino acids. {Q&A}
Small PEPTIDES [I emphasize peptides here, that is, oligopeptides, not polypeptides], like the sub-peptides produced by trypsin digestion of the polypeptide pictured above, can also be separated by both of these techniques; the properties of the peptides will be a COMPOSITE of the properties of the constituent amino acids. That is, each of the amino acid side groups in a particular peptide will contribute some hydrophobicity or charge or polar quality; the resulting peptide will reflect the combination of all these effects. For example, a peptide with 2 arginines and one glutamic acid as the only charged residues will have a net charge of +1 at pH7 and so migrate toward the cathode in paper electrophoresis. {Q&A}
To review chromatography, try problems 2-9 A & B & 2-10.
One of the most famous examples of the use of these
methods to analyze peptides rather than single amino acids was in the study of
sickle cell disease, a genetic disease. Sickle cell disease is caused by an abnormal hemoglobin
protein and results in abnormally shaped red blood cells that can clog
capillaries and so prevent blood flow. Hemoglobin is made up of several components, one of which is a
polypeptide called beta-globin. The sequence of amino acids in beta-globin from
sickle cell hemoglobin was found to differ from that of normal beta-globin. In
fact, the difference between sickle cell beta-globin and normal beta-globin was
the first case i which the molecular basis of a genetic disease was defined.
instance in which the by Vernon Ingram in the 1960's. Ingram chopped up the sickle cell beta-globin
into small peptides. This digestion can be done using enzymes
(endo-proteases, that cleave polypeptides in a specific way, that is, at
specific amino acid residues. For example, as mentioned above, the
protease trypsin hydrolyzes polypeptides after (i.e., on the carboxyl side of)
lysine and arginine. So first treat the beta globin protein with trypsin
to break it into pieces (sub-peptides) by hydrolysis after lys and arg residues.
The resulting mixture of sub-peptides (outside of this lecture they are just called peptides, the "sub" being understood from the context) are then first separated along one edge of a filter paper sheet by paper electrophoresis. Note that these are peptides that are migrating, not free amino acids. The sheet is then turned 90 degrees and subjected to paper chromatography.
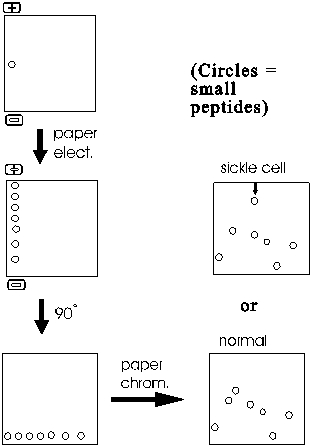
The result is a series of spots (after staining to visualize their positions) representing all the sub-peptides.
One peptide migrates differently in sickle cell globin compared to normal globin. This peptide can then eluted from the paper and sequenced. Comparison with the normal counterpart peptide shows that the sickle cell globin carries a single amino acid substitution. In place of glutamic acid, it has a valine at one position in the peptide. How could such a single change have such a large effect? The answer lies in the 3-dimensional shape of proteins, to which we will soon discuss.
Most proteins can be separated into characteristic patterns of spots this way. No two proteins have the same primary sequence, and so each protein will yield a different set of sub-peptides after trypsin digestion. Most of the sub-peptides from any two polypeptides will migrate differently, so the total pattern of spots will be different for each protein. The procedure is called FINGERPRINTING a protein, since the migration patterns are so characteristic. I use the term fingerprinting to refer to the entire process of digesting the polypeptide into sub-peptides, separating it in 2 dimensions and then visualizing and comparing the spots. {Q&A}
To review fingerprinting, try problem 2-11.
Protein 3-dimensional structure
Now let us return to polypeptide structure.
Each polypeptide has a particular sequence of amino acids. Thus if we could examine several molecules of the protein albumin we might find:
Molecule #1: N-met-leu-ala-asp-val-val-lys-....
Molecule #2: N-met-leu-ala-asp-val-val-lys-...
Molecule #3: N-met-leu-ala-asp-val-val-lys-... etc.
So they have the same primary structure. But as always, we must consider structure in 3-dimensional space for a real picture of the molecule.
While the linear structure is the same, the 3-D structure for each molecule must surely be different in solution, no? After all, thermal motion will be buffeting this rope of strung-together amino acids all about, so that each molecule will be expected to take on a random configuration, no? Look at this scale model of a POLYPEPTIDE OF 500 amino acids, a CLOTHES LINE. The dimensions are about right, but the side chains have been left out. I have put colored parts of the rope red to indicate polar side chains, the white parts being apolar or hydrophobic [board]. At 37 degrees, you might imagine this clothesline in a Jacuzzi, constantly taking on new shapes, with its hydrophilic side chains constantly forming new hydrogen bonds to water.
This is the wrong picture. A more appropriate picture is a bundled up rope, folded into a compact structure that withstands this thermal motion at body temperatures [bundled rope].. red on outside ...white hydrophobic on inside (which makes sense based on the weak bond behaviors we discussed).
OK, maybe this molecule could collapse on itself .. after all the hydrophobic side chains will tend to aggregate. But if we took another molecule, another linear chain, it would probably fold a different way, after all, 500 amino acids, there must be many many ways to get the hydrophobics inside. I could stuff the white parts of the rope together and put them on the inside in many different ways: leucine-2 pushed up against valine 25 in one molecule but associated with leucine-346 in the next molecule, so we might expect each of these protein molecules to have a unique structure in 3-D space:
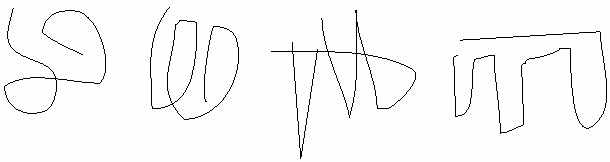
But in fact if we look for a second folded up example of this
molecule, it looks like this [second rope bundle], exactly the same
as the first (note loop count, etc.). Protein molecules exist as precisely
defined 3-dimensional structures in solutions, each molecule like the next,
super imposable.
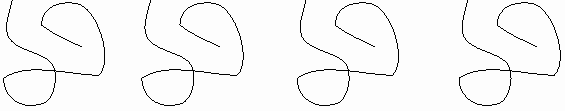
That is, a typical polypeptide chain, having some 10,000 atoms linked together, is folded up so that these 10,000 atoms all have the same position relative to each other in each and every molecule you examine. This still amazes me. How could this be? How could each molecule find just the right structure as it folds up, each and every time? Let's see.
Well, what is holding the molecule in this shape? The four weak bond types we discussed earlier, plus one new bond to be described in a few minutes.
So now we can see that one polypeptide molecule can be folded into a compact structure and we can understand what holds it together, but why is it that there is only one structure formed and not many? Is there only one solution to the folding problem for a particular polypeptide chain?
Perhaps all possible conformations are tried in the course of folding, and only the most stable one accumulates. Can we predict the conformation from first principles? If we plug in the properties of all the amino acid side chains, how hydrophobic they are, what is the strength of an ionic bond, etc. we can ask a computer to try and try many combinations, many interactions. This is a very difficult computer problem, even for today's supercomputers, because the number of possibilities for a good-size polypeptide of say 500 amino acids is enormous (20^500). But it has been tried, and so far usually the wrong structure comes out if one relies solely on first principles. (more accurate predictions can be made by comparing an unknown protein to known examples). The right structure is determined by examining crystals of the proteins, beaming X-rays through the protein crystals and calculating how they are refracted by the atoms in the crystal. Perhaps we really don't know the right properties of the side chains. Or perhaps there is some guide to folding that being imposed on the polypeptide as it is being polymerized in the cell, some outside influence, even a template of sorts, one can imagine a plaster mold analogy, for example. That is not the case, as we shall see next.
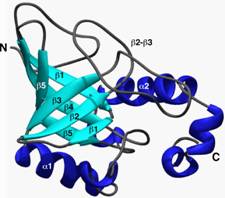
(Denaturation,
renaturation)
Well, if it is true that the folded
structure of a particular protein is unique simply because it is the most
stable, then if we UN-fold the polypeptide, it should be able to RE-fold into a
its unique structure. How could we unfold a protein, let's say one with only weak bonds
holding the protein i its 3-D shape. We could consider egg albumin, for an
everyday case of polypeptide denaturation. Raw egg white is a concentrated
solution of this single ~500 amino acid polypeptide, that exists folded into a
roughly spherical shape.
How can we denature it?....The 3-D structure is being held together in most proteins lust by the weak bonds we have discussed (H-bonds, ionic bonds, van der Waals bonds and hydrophobic forces). Heating proteins typically unfolds them: thermal motion becomes too great for the weak bonds.
Let's heat it to denature it (boil it). The sphere is now subject to faster and faster thermal motions, until finally it starts to unravel. What has happened to the egg white (the albumin polypeptide)? It has become denatured. Purves6ed 3.9; 7th 3.11]. No longer native, which is the structure in the cell. No covalent bonds have been broken by this 100oC temperature. The bundled up rope became the open randomly coiled rope in the Jacuzzi, and this allowed many wrong bonds to form, it exposed the hydrophobic groups normally hidden in the interior of the protein. In this concentrated solution a tangled mass of interacting polypeptide chains was produced, which resulted in a gel, a hot hard-boiled egg. So while folded up polypeptides are stable enough in their native environment inside the cell, the 3-dimensional structure is typically rather fragile: most proteins are easily denatured by heat and other treatments that can affect these weak bonds. This bundled rope in the Jacuzzi exists on the verge of becoming unraveled.
So now let's reverse the denaturation, let's cool down this hard-boiled egg and return it to normal temperatures. The gel seems to stay. We do not get back our runny egg white. A case of irreversible denaturation. But not a very fair experiment, letting all those molecules, present at a very high concentration, get SO tangled with each other. Let's try a denaturation - renaturation in a more gentle, gradual way.
A fellow named Christian Anfinsen did this experiment in the 1950's. He took a protein called ribonuclease, a protein that is a digestive enzyme, a protein that helps break down the macromolecule RNA. It must be in its native folded structure to do this job.
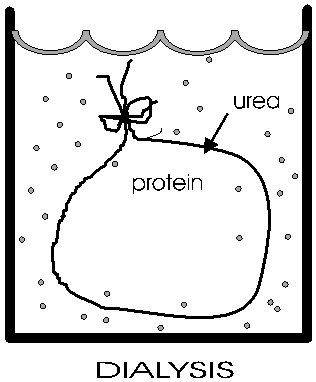
(Dialysis)
Anfinsen placed the polypeptide in a plastic sack, and added urea,
H2N-CO-NH2
to the solution outside the sack. At high concentrations (e.g., 8M), urea will break hydrogen bonds {Q&A} {Q&A} The sack is made of a semi-permeable plastic material with pores big enough to allow small molecules like urea and water to pass through but not macromolecules like albumin or ribonuclease. This process of allowing the concentrations of small molecules to change while holding the concentrations of large molecules constant is a called dialysis. After allowing time for diffusion, the concentration of urea inside the sack should be the same as the concentration outside. {Q&A} He then checked that the protein had become denatured (e.g., by ultracentrifugation, see below). And it no longer do its job catalyzing the hydrolysis of RNA.
Now he gradually dialyzed out the urea (by changing the solution outside the sack to stepwise lower and lower concentrations of urea). A dilute solution of the protein was used, and the gradual removal of the urea gave time for the polypeptide to re-fold.
He got back native ribonuclease. It checked out physically, and also functionally, by the fact that it regained its ability to digest RNA.
This type of experiment has been now been repeated many times for many different proteins. It works for many, fails for some. But the positive results are very important, for they prove that for many or even most proteins, all the information that is necessary for the complex and unique 3-dimensional structure is present in the primary sequence of the polypeptide chain.
The discovery of this fundamental tenet of biochemistry earned Anfinsen the Nobel Prize.
© Copyright 2010 Lawrence Chasin and Deborah Mowshowitz Department of Biological Sciences Columbia University New York, NY
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[Purves 6:3.20, 7:3.19]](http://www.columbia.edu/cu/biology/courses/c2005/purves6/figure03-20.jpg){kind=link}
![[Purves 6:3.21, 7:3.20]](http://www.columbia.edu/cu/biology/courses/c2005/purves6/figure03-21.jpg){kind=link}
![[Purves 6:3.22, 7:3.21]](http://www.columbia.edu/cu/biology/courses/c2005/purves6/figure03-22.jpg){kind=link}
{kind=link}
{kind=link}
![[Purves 6:5.1, 7:5.1]](http://www.columbia.edu/cu/biology/courses/c2005/purves6/figure05-01.jpg){kind=link}
{kind=link}
{kind=link}
![[Purves6ed 2.21a]](../purves6/figure02-21a.jpg){kind=link}
![[Purves6ed 2.21b]](../purves6/figure02-21b.jpg){kind=link}
{kind=link}
![[Purves6ed 3.4]](../purves6/figure03-04.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
![Purves6ed 3.9; 7th 3.11]](../../c200508/lectures/purves6/figure03-09.jpg){kind=link}