![[Purves6ed 2.21a]](../purves6/figure02-21a.jpg){kind=link}
![[Purves6ed 2.21b]](../purves6/figure02-21b.jpg){kind=link}
(With embedded Q&A)
Lec. 4. Biol C2005/F2401 2005 L. Chasin
Sept. 14, 2006
Last updated:
Thursday, September 14, 2006 12:19 AM
© Copyright 2006 Lawrence Chasin and Deborah Mowshowitz
Department of Biological Sciences Columbia University
New York, NY
Handout: Use last time's
Props: 2 aa
models with extra hydrogens, 3 ropes, wire alpha helix, wire 3-D protein
structure
Proteins cont.
Stereoisomers: L and D amino acids
Protein structure (4
levels):
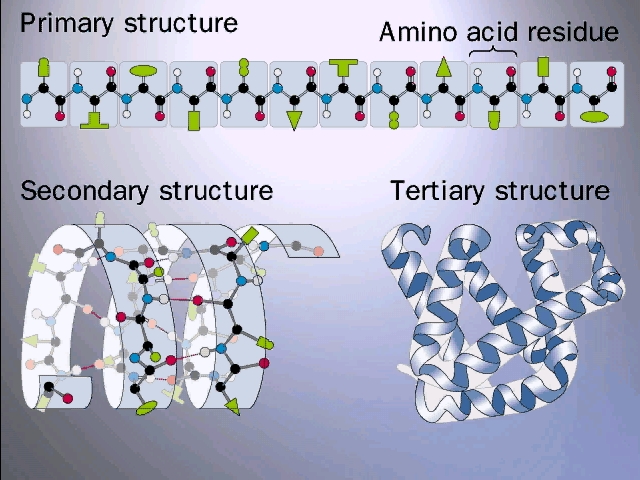
Primary (1o)= linear sequences of AAs
Polypeptides, peptide bond, backbone, N-terminal, C-terminal
Methods:
Paper electrophoresis of amino acids
Paper chromatography of amino acids
Fingerprinting: 2-dimensional electrophoresis + chromatography of small peptides
Sickle cell disease: an altered sub-peptide of the globin polypeptide
Primary (1o) structure = AA seq
Constrained rotation of the backbone
Secondary (2o)= alpha-helix, beta sheet
Tertiary(3o) = overall 3-D, side chain interaction
(Stereoisomers)
Now let's consider the structure of an amino acid in
3 dimensions:
When carbon forms 4 single bonds, it makes them spaced equally apart from each other in space, in the form of a tetrahedron as in this representation of glycine [a model with 2 white groups (the H's) is shown].
Now consider this other molecule of an amino acid [again with 2 white groups], with 2 H's of glycine, e.g. Are these the same molecule, that is, are they distinguishable or are they indistinguishable?
They are indistinguishable, since I can rotate them and superimpose their atoms.
But now suppose I make this alanine instead of glycine. I replace one H with a [orange] -CH3 group on each molecule [I am being sure to make them stereoisomers].
I can no longer superimpose them. They are both alanine, as they have the same four groups attached to the central carbon. But in three dimensions they are actually mirror images of each other. See [Purves6ed 2.21a]. We call one D-ala and one L-ala. See [Purves6ed 2.21b].
This one is D, or is it this one... ? I can't remember .. it's not too important here.
What is important is that in general, you have this situation, the possibility of two stereoisomers, whenever there is what is called an ASYMMETRIC carbon atom in a molecule, that is, a carbon with four different groups attached.
These stereoisomers are sometimes called optical isomers, since the two forms, in solution, will bend a beam of polarized light one way or the other. Thus the D designation originally meant dextro, or to the right, whereas L stood for levo, to the left.
All amino acids except glycine have an asymmetric carbon, which is the alpha-carbon. So we can draw 19 of the amino acids in 2 stereoisomeric forms.
So do we really have 39 a.a.'s? No. All the stereoisomeric forms of the amino acids in proteins are L-amino acids, so we only have to worry about 20.
Note that the sugars we discussed, like glucose, have several asymmetric carbon atoms. Aside from L and D designations, the sugar stereoisomers are given different English names (e.g., D-glucose, D-mannose, L-rhamnose, etc.).
To review the various chemical groups discussed so far, try problem 1-19.
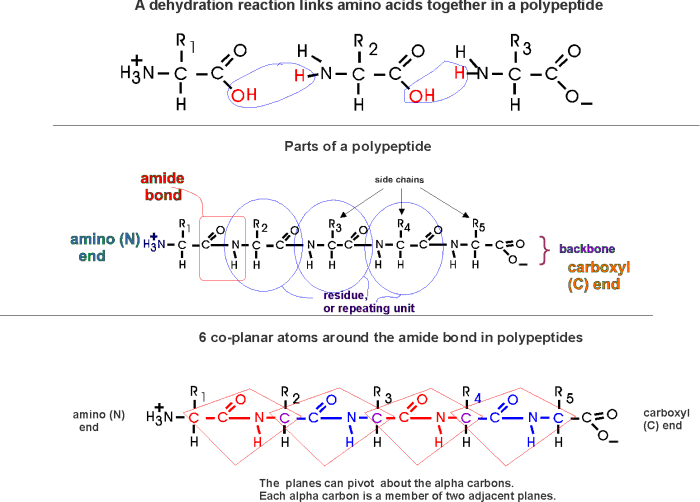
(Polypeptides, peptide bond)
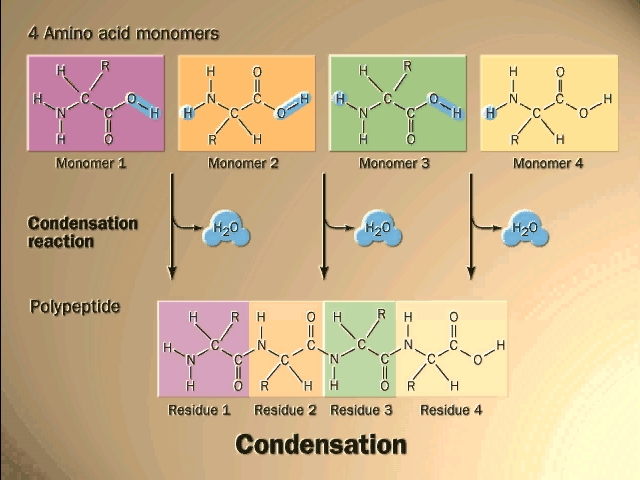
Polymerization of aa's
OK, now let's now string these L-amino acids together, polymerize them. The
bond that connects two amino acids is an AMIDE bond
(-CO-NH-) between the carboxyl of one amino acid and the amino group of the
next. Once again, a molecule of water is removed in the formation of the
connecting bond:
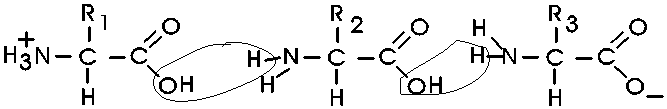
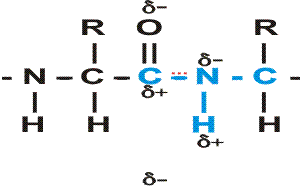
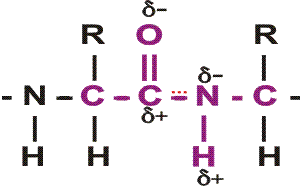
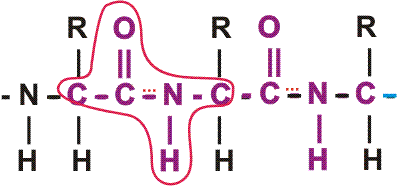
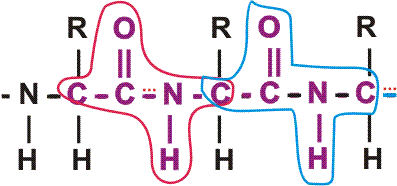
In the special case of proteins, this amide bond is called a PEPTIDE BOND, and the resulting product a PEPTIDE, a dipeptide (or we could go on to a tri-peptide, oligo-peptide, or finally, POLY-PEPTIDE). (See also polypeptide handout). Also see [Purves6ed 3.4], and another picture. {Q&A}
By convention, the amino group is written on the left for an amino acid and also for a peptide.
In the tripeptide in the diagram, note the peptide bond (boxed), and the repeating unit, or aa "residue" [circled]. Residue refers to what's left of the amino acid monomer after it has been incorporated into a polypeptide, which is most of it: it just lacks one H at what was the amino end and one OH at what used to be the carboxyl end. Note also that the charged amine and carboxyl groups no longer exist inside the polypeptide, having been replaced by the amide, an uncharged (but polar) functional group.
Almost all polypeptides have 2 ends, the amino end and the carboxyl end, which do remain charged at pH7.

The "backbone" of the polypeptide is defined as all of the atoms except the side chains.
The only free amino and carboxyl atoms of the backbone are at the 2 ends.
The side chains , then, stick out of this backbone (also see polypeptide handout).
Nomenclature: e.g., alanine-methionine-alanine, or ala-met-ala, or alanyl-methionyl-alanine, or AMA
To review peptide structure, try problem 1-15, C, and then try 2-3 part A.
The length of polypeptides is commonly 100-1000 amino acids, but smaller and larger ones also can be found.
Each and every protein molecule in the cell has an identity defined by its particular sequence of amino acids. Each E. coli cell contains about 3 million polypeptides molecules, but only about 3000 different ones. Each of these individual protein types has a name to go along with its chemical identity.
Are there enough combinations to specify 3000 different polypeptide sequences? Well, if the average polypeptide is 500 amino acids long, then the possible combinations are 20500 or 10650, which is a number of inconceivable magnitude. So evolution has settled on about 100,000 of these combinations to do biological jobs.
Some examples of polypeptides, taken not from E. coli, but from more familiar organisms include:
Carriers: hemoglobin, which carries oxygen in red blood cells;
Nutrients: egg albumin, a nutrient in the white of a hen's egg;
Structural: keratin, providing toughness in skin, fingernails, and wool;
collagen, providing a strong connection between cells in tendons;
Signal reception: estrogen receptor (intracellular)
epidermal growth factor receptor (spanning the cell membrane)
Recognition of foreign substances: immunoglobulins (antibodies)
Enzyme catalysts: beta-galactosidase, which helps digest the milk sugar lactose.
We will discuss enzymes in some detail as an important category of proteins.
(Primary (1o)
structure = linear
sequences of AAs)
Each of these proteins contains a polypeptide with a particular sequence of
amino acids, usually all 20 are represented, although not at all equally. Unlike
polysaccharides, this sequence usually exhibits no obvious simple regularity, or
repeating subsequence:
This linear sequence of amino acids is called the primary (1o) structure of a protein. {Q&A}
It might be, for instance: met-ala-leu-leu-arg-glu-leu-val ...... How is this sequence determined?
(Methods: Paper chromatography,
electrophoresis, fingerprinting)
I will discuss now a bit of some
methodology used in the purification of amino acids and proteins. We
bring in some selected lab methods from time to time for two reasons: First, the
behavior of molecules in experimental situations helps you to understand their
behavior in nature; and second, the methodology is interesting in its own right
as an example of how science is done.
Our first topic of methodology is directed at the question of how we get to know this primary structure, this sequence of amino acids in a polypeptide?
The modern way to determine the sequence of a polypeptide involves an instrument called a mass spectrometer: the protein is fragmented and the exact molecular weight of each small fragment is determined by its flight path in an electric field in the instrument. From repetitions of this type of analysis the exact amino acid sequence can be deduced.
A more traditional method used to determine the sequence of a polypeptide is to chemically degrade from one end it in a stepwise fashion, starting at either the amino or the carboxyl end. First you must purify the polypeptide in question away from the other 3000 polypeptides in the cell; we will discuss that process a little later.
The degradation of the polypeptide back to its free monomer AA's is a form of HYDROLYSIS, a reverse of the dehydration that accompanied the formation of the peptide bond. As an example we will only discuss here the degradation of a peptide or polypeptide from the C-terminal. The controlled hydrolysis of amino acid residues from the carboxyl end of a polypeptide is a form of enzymatic hydrolysis; an enzyme, called carboxypeptidase, itself a polypeptide, catalyzes this hydrolysis; it does not happen by itself. Carboxypeptidase is called an exopeptidase since it works on the end (exterior) of the polypeptide. We will learn more about enzymes next week. After the carboxypeptidase is mixed with a peptide and hydrolysis begins: all the trillions of molecules release their C-terminal amino acid in unison, almost synchronously, so that in the first wave the last (original C-terminal) amino acid is released. A bit of the reaction mixture is remove at this point the released amino acid is separated from the main peptide and identified. By letting the reaction proceed for increasing amounts of time, the time that amino acids are released can be correlated with their distance form the C-terminal end.
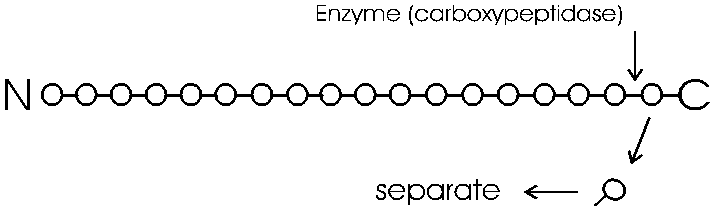
You can get the sequence of perhaps 20 amino acids in from the carboxy terminal in this way, before the process breaks down. Since most polypeptides are greater than 20 amino acids in length, you first need to chop the polypeptide into manageable pieces and then sequence each piece by subjecting it to hydrolysis by carboxypeptidase. This internal chopping is done using a different type of proteolytic enzyme, an endopeptidase (cleaves inside, not from the end). One such enzyme is trypsin, which cleaves after the 2 amino acids with basic side groups, arginine and lysine.
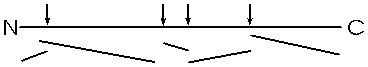
Let's consider the analysis of the sequence of one of these sub-peptides produced by trypsin cleavage. The problem is how to separate and identify the different amino acids that are released by this carboxypeptidase hydrolysis. How do you know which amino acid came off when? Amino acids behave sufficiently differently from each other under certain conditions to allow the complete separation of all 20 species from a mixture. We will discuss two methods for separation and identification here.
One way is based on the migration of amino acids in an electric field. In PAPER ELECTROPHORESIS, an amino acid mixture is spotted onto a sheet of filter paper, the paper is wet with a buffer salt solution and placed between two electrodes and high voltage (e.g., 2000 volts) applied. At neutral pH, the acidic amino acids (asp and glu) will have a net negative charge and will migrate toward the ANODE (+ pole) while the basic amino acids (arg and lys) will migrate toward the CATHODE (- pole). {Q&A} Electrically neutral amino acids will not migrate much, unless the pH is made acidic or basic (as it is in some problems in the problem book) .
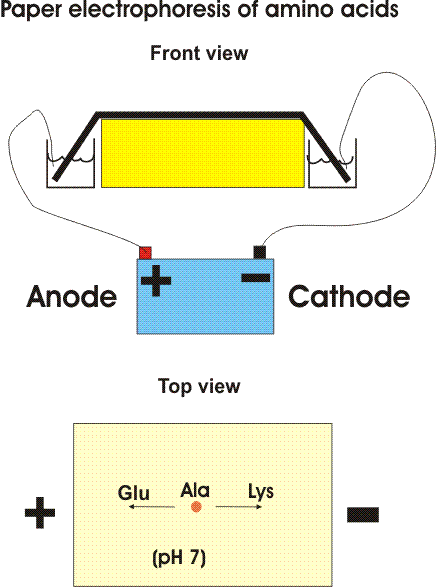
Viewed after application in the center followed by electrophoresis.
Try problem 2-1.
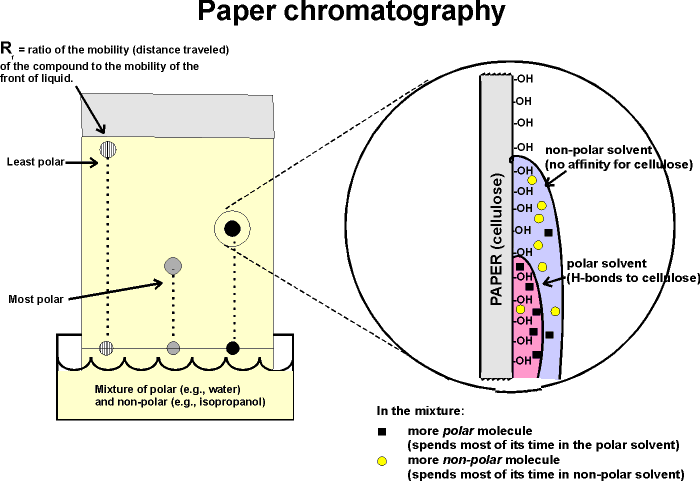
A more versatile separation method is PAPER CHROMATOGRAPHY. This method is based on the differential solubility of the different amino acids
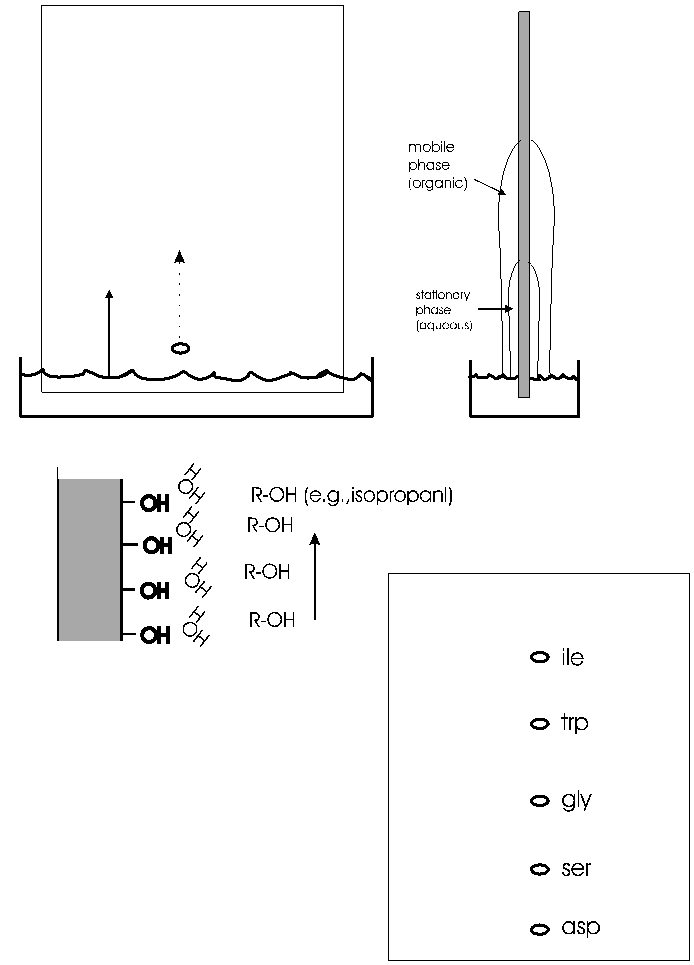
in organic (non-polar) solvents, which in turn is determined by the nature of the side group. The amino acid mixture is spotted onto a filter paper; one edge of the paper is immersed in a mixture of aqueous and non-aqueous solvents. (See handout.) The liquid will be drawn up the paper by capillary action. As it rises, the water in the liquid mixture is bound by the paper (cellulose, with its many OH groups), forming a stationary water layer, or stationary phase. The organic solvent (e.g., propanol) moves up without as much interaction with the solid cellulose; it is considered the mobile phase. The amino acids will be constantly equilibrating between being in the mobile organic phase or the stationary water phase. The more polar the side chain, the more time the amino acid will spend in the stationary phase. The more hydrophobic the side chain, the more time it spend in the mobile organic phase. By using a series of different solvents, all 20 amino acids can be separated in this way. It works for many other organic molecules as well. The distance that an organic molecule moves in a particular chromatographic system is called the Rf, which stands for mobility Relative to the Front, that is, the distance the organic molecule in question migrated divided by the distance that the front of liquid has risen on the paper at that time. Rf's in a particular solvent and at a given temperature are reproducible and are published for many organic compounds, including all the amino acids. {Q&A}
Small PEPTIDES [I emphasize peptides here, that is, oligopeptides, not polypeptides], like the sub-peptides produced by trypsin digestion of the polypeptide pictured above, can also be separated by both of these techniques; the properties of the peptides will be a COMPOSITE of the properties of the constituent amino acids. That is, each of the amino acid side groups in a particular peptide will contribute some hydrophobicity or charge or polar quality; the resulting peptide will reflect the combination of all these effects. For example, a peptide with 2 arginines and one glutamic acid as the only charged residues will have a net charge of +1 at pH7 and so migrate toward the cathode in paper chromatography. {Q&A}
To review chromatography, try problems 2-9 A & B & 2-10.
One of the most famous examples of the use of these
methods to analyze peptides rather than single amino acids was in the study of
sickle cell disease. Sickle cell disease is caused by an abnormal hemoglobin
protein. Hemoglobin is made up of several components, one of which is a
polypeptide called beta-globin. The sequence of amino acids in beta-globin from
sickle cell hemoglobin was found to differ from that of normal beta-globin. That difference
was determined (by Vernon Ingram in the 1960's) by chopping up the sickle cell beta-globin
into small peptides. This digestion can be done using enzymes
(endo-proteases, that cleave polypeptides in a specific way, that is, at
specific amino acid residues. For example, as mentioned above, the
protease trypsin hydrolyzes polypeptides after (i.e., on the carboxyl side of)
lysine and arginine. So first treat the beta globin protein with trypsin
to break it into pieces (sub-peptides) by hydrolysis after lys and arg residues.
The resulting mixture of sub-peptides (outside of this lecture they are just called peptides, the "sub" being understood from the context) are then first separated along one edge of a filter paper sheet by paper electrophoresis. Note that these are peptides that are migrating, not free amino acids. The sheet is then turned 90 degrees and subjected to paper chromatography.
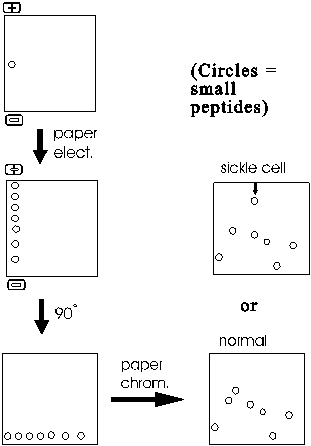
The result is a series of spots (after staining to visualize their positions) representing all the sub-peptides.
One peptide migrates differently in sickle cell globin compared to normal globin. This peptide can then eluted from the paper and sequenced. Comparison with the normal counterpart peptide shows that the sickle cell globin carries a single amino acid substitution. In place of glutamic acid, it has a valine at one position in the peptide. How could such a single change have such a large effect? The answer lies in the 3-dimensional shape of proteins, to which we will turn next.
Most proteins can be separated into characteristic patterns of spots this way. No two proteins have the same primary sequence, and so each protein will yield a different set of sub-peptides after trypsin digestion. Most of the sub-peptides from any two polypeptides will migrate differently, so the total pattern of spots will be different for each protein. The procedure is called FINGERPRINTING a protein, since the migration patterns are so characteristic. I use the term fingerprinting to refer to the entire process of digesting the polypeptide into sub-peptides, separating it in 2 dimensions and then visualizing and comparing the spots. {Q&A}
To review fingerprinting, try problem 2-11.
Protein 3-dimensional structure
Now let us return to polypeptide structure.
Each polypeptide has a particular sequence of amino acids. Thus if we could examine several molecules of the protein albumin we might find:
Molecule #1: N-met-leu-ala-asp-val-val-lys-....
Molecule #2: N-met-leu-ala-asp-val-val-lys-...
Molecule #3: N-met-leu-ala-asp-val-val-lys-... etc.
So they have the same primary structure. But as always, we must consider structure in 3-dimensional space for a real picture of the molecule.
While the linear structure is the same, the 3-D structure for each molecule must surely be different in solution, no? After all, thermal motion will be buffeting this rope of strung-together amino acids all about, so that each molecule will be expected to take on a random configuration, no? Look at this scale model of a POLYPEPTIDE OF 500 amino acids, a CLOTHES LINE. The dimensions are about right, but the side chains have been left out. I have put colored parts of the rope red to indicate polar side chains, the white parts being apolar or hydrophobic [board]. At 37 degrees, you might imagine this clothesline in a Jacuzzi, constantly taking on new shapes, with its hydrophilic side chains constantly forming new hydrogen bonds to water.
This is the wrong picture. A more appropriate picture is a bundled up rope, folded into a compact structure that withstands this thermal motion at body temperatures [bundled rope].. red on outside ...white hydrophobic on inside (which makes sense based on the weak bond behaviors we discussed).
OK, maybe this molecule could collapse on itself .. after all the hydrophobic side chains will tend to aggregate. But if we took another molecule, another linear chain, it would probably fold a different way, after all, 500 amino acids, there must be many many ways to get the hydrophobics inside. I could stuff the white parts of the rope together and put them on the inside in many different ways: leucine-2 pushed up against valine 25 in one molecule but associated with leucine-346 in the next molecule, so we might expect each of these protein molecules to have a unique structure in 3-D space:
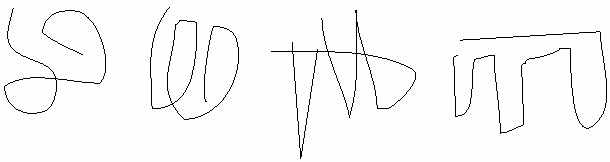
But in fact if we look for a second folded up example of this molecule, it looks like this [second rope bundle], exactly the same as the first (note loop count, etc.). Protein molecules exist as precisely defined 3-dimensional structures in solutions, each molecule like the next, super imposable.
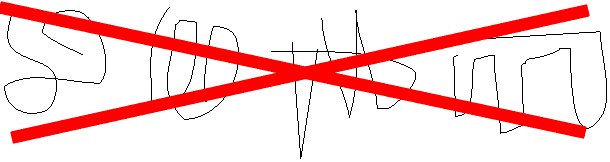
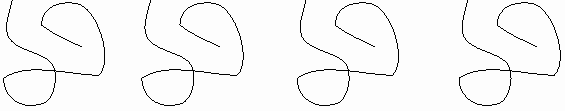
That is, a typical polypeptide chain, having some 10,000 atoms linked together, is folded up so that these 10,000 atoms all have the same position relative to each other in each and every molecule you examine. This still amazes me. How could this be? How could each molecule find just the right structure as it folds up, each and every time? Let's see.
Well, what is holding the molecule in this shape? The four weak bond types we discussed earlier, plus one new bond to be described in a few minutes.
Let's consider how this folding looks in more detail:
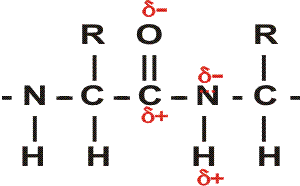
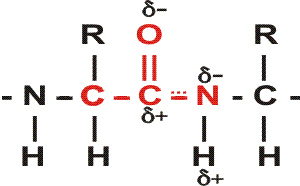
First, the flexible rope was not a good representation of even the backbone, because the peptide bond itself imposes some constraint on structure. The peptide bond itself has a property that influences all polypeptides regardless of the side chains. Because of the electronegativity difference between C and O or N, there is a partial separation of charge, one you could have predicted.
What you may not have realized is that the partial + charge on the C and the partial - charge on the adjacent N, imparts a partial extra bond between those 2 atoms, and thus a partial double bond character to the C-N bond. This partial double bond is sufficient to stop free rotation about the C-N bond. Thus the backbone is not free to rotate around all connections, but rather each repeat contains 6 atoms confined to one plane:
[The four red atoms (third figure) lie in one plane. The four blue atoms (fourth figure) lie in one plane. The 4 red atoms and the 4 blue atom have 2 atoms in common (the C-N), both of which lie in the same plane. So all six atoms must lie in the same plane]
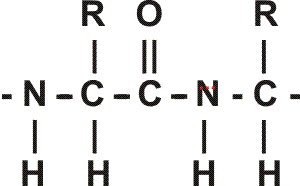
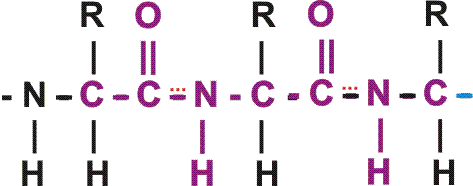
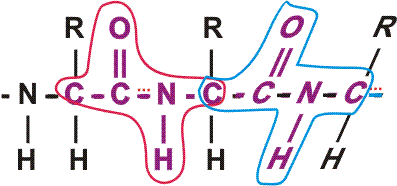
The polypeptide can be visualized as having a series of planes, each able to rotate about one another. So a chain would be a better representation than a rope. See handout.
(Secondary (2o)=
alpha-helix, beta sheet)
This partial separation of charge also means that the O and the NH of the peptide
bond can hydrogen bond... to water for example. Since the NH is a hydrogen donor
and the O is a hydrogen acceptor for a hydrogen bond, we should consider the
possibility that these groups can H-bond to each other. But H-bonds require
a linear orientation of the 3 atoms involved, so certainly the NH of the very
next residue cannot H-bond to a C=O preceding it. But what about the next residue?
No, still can't make it. But by the fifth residue down you are able to line
up an NH to the O: -C=O..H-N-. i.e., there are 3 complete
residues 3 in between.
{Q&A}


So the C=O of #1 can H-bond to the HN of #5. But then also the C=O of #2 should be able to H-bond to the HN of #6, and so on. This twisting and H-bonding can hold the backbone in a HELIX, the so-called alpha-helix. {Q&A}
The alpha-helix is an example of secondary structure, which is (my definition): structure produced by regular repeated interactions between atoms of the backbone.
We might expect all the amino acid backbone atoms to be in an alpha-helical conformation, but we have left out consideration of the side chains, which can greatly influence the folding, as we will see in a minute.
The alpha-helix is not the only form of secondary structure, there is another, the beta-pleated sheet. In this case we once again have the C=O and the NH of the backbone forming H-bonds to each other, but in this case two sections of the polypeptide are aligned side by side (each vertical line represent a different region of the same polypeptide):
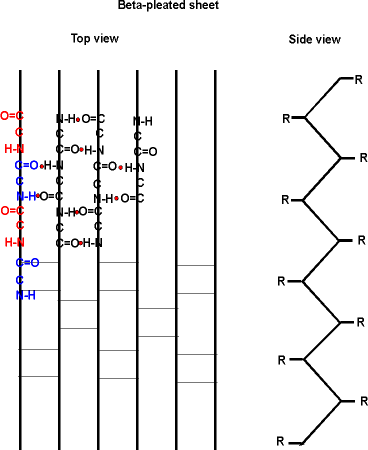
Several sections of polypeptide can line up like this, to produce a sheet of strands. The chains are usually anti-parallel, but parallel alignments are also possible.
See your texts for better pictures of these structures. B: 49 and Purves6ed 3.5a.
Once again, side chain interactions play a major role in allowing or disallowing such secondary structures to form. But in fact, most proteins do have extensive regions folded into alpha-helices and beta-pleated sheets.
Secondary structure consists mostly of these 2 structures. {Q&A}
To review secondary structure, try problem 2-3 part C.
(Tertiary
(3o)= overall 3-D of a polypeptide)
Tertiary structure means
the overall 3-dimensional folding of a single polypeptide chain. For this
overall shape, interactions between side chains are very important, as are
interactions between side chain and water. A generality is that
the hydrophilic groups are folded to be on the outside where they can interact
with water via hydrogen bonds, while the hydrophobic side chains are collected
in the inside of the structure, pushed together by hydrophobic forces. This rule
is not at all 100% true, and most proteins have side chains that deviate from
this generality. That is, there are hydrophobic side chains on the surface, but
they are intermingled among the hydrophilic groups. And there are hydrophilic
groups on the inside, where they are usually interacting with other hydrophilic
groups. In fact, it is this interaction of side chains with each other that
confers most of the overall 3-dimensional shape on a given polypeptide.
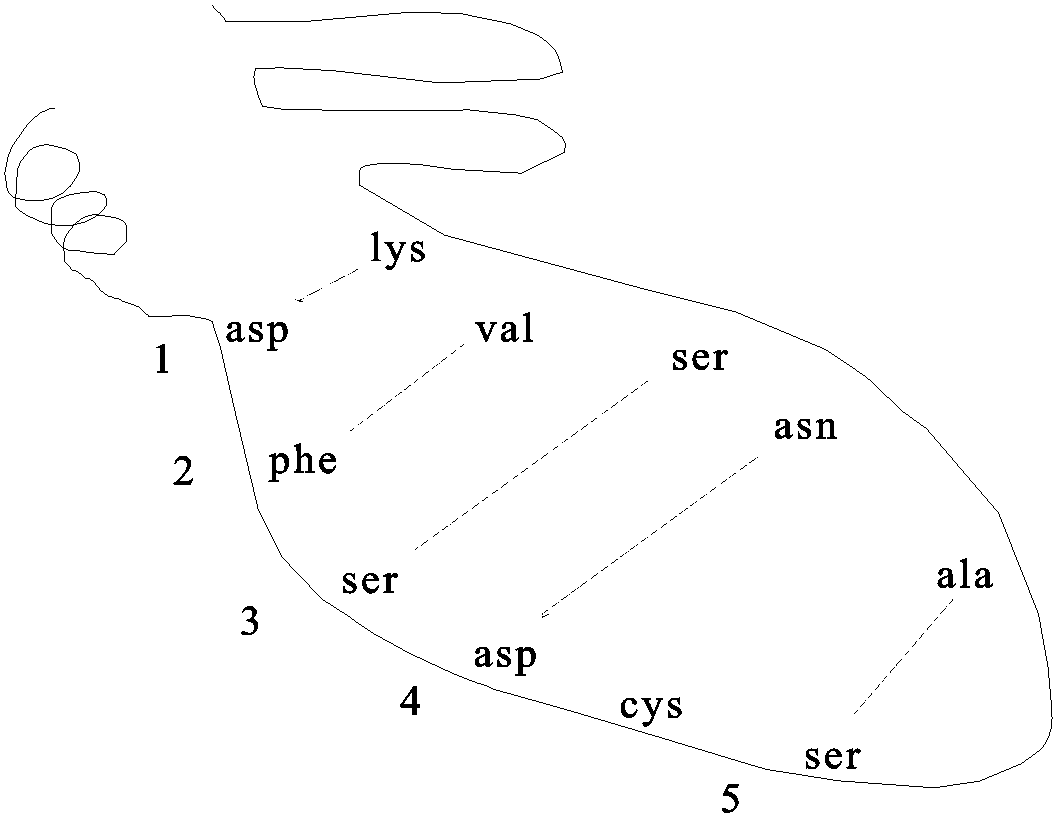
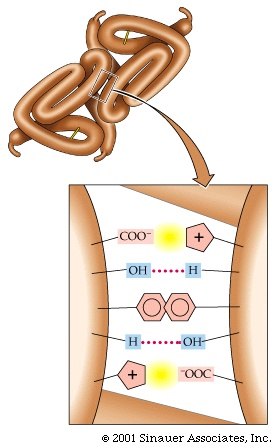
Pictured here are the weak bonds that were introduced earlier. The side chain interaction indicated in the diagram illustrate examples of these various interactions. Consult your text for the exact nature of the side chains:
1. ionic (lys - asp)
2. hydrophobic and VDW (phe - val)
3. H-bond (ser - ser)
4. H-bond to ionic (asp - asn)
5. van der Waals (ser - ala)
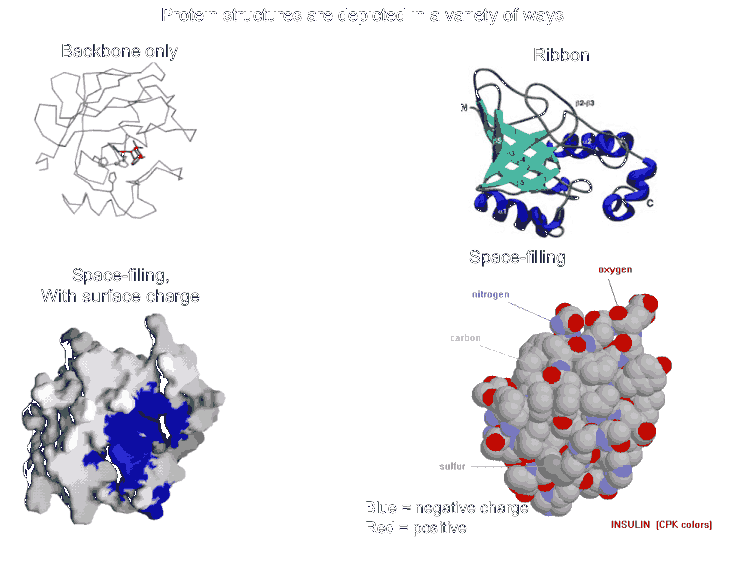
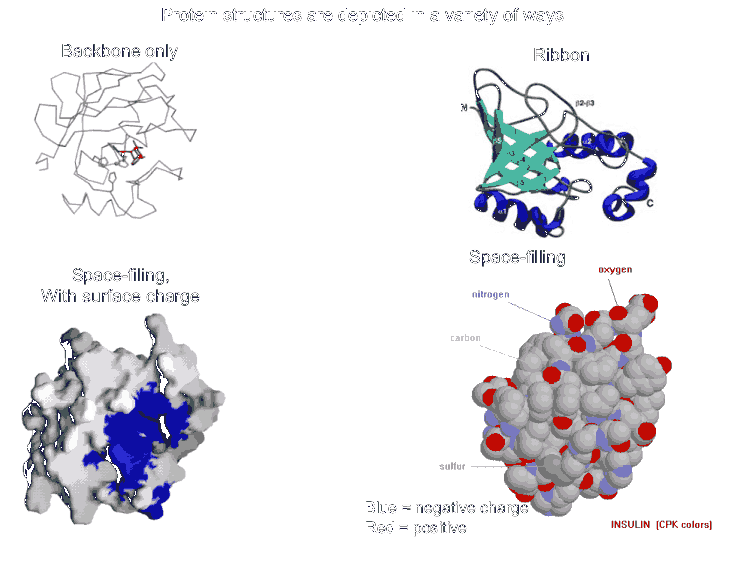
Most proteins fold into a roughly globular shape (enzymes, Hb, antibodies--see pictures of proteins in your texts: a space-filling model, or showing just the backbone connections, or a ribbon model), but many take on an elongated or even fibrous shape (collagen, myosin [in muscle], fibroin [in silk]).
These are weak bonds, but in the aggregate, they are strong enough to hold the polypeptide together at least under the thermal motion conditions of physiological temperature (37 deg C) (Purves6ed 3.8). {Q&A}
(Sulfhydryls,
disulfides)
But there is one strong bond that contributes to the folding of some proteins.
This is the DISULFIDE BOND, and it differs from all these other bonds in being a
covalent bond. It can only be formed between the side chains of two CYSTEINE
residues. The side chain -CH2-SH contains a SULFHYDRYL group: -SH. Two
sulfhydryls can react with oxygen to lose their 2 hydrogen ATOMS (H with
its electron; not H+ ions, not protons) and become bound to each other in the
process:
Protein-CH2-SH + HS-CH2-Protein + ˝O2 ---> Protein-CH2-S-S-CH2-Protein + H2O
So now the two sulfur atoms are sharing electrons in a strong covalent bond. This bond cannot be broken by mere thermal energy, and so disulfide bonds hold the two parts of the polypeptide chains that had contained the two cysteines firmly together. Not all proteins have disulfide bonds, but many do.
This reaction is an example of an oxidation-reduction reaction: the sufhydryls are getting oxidized (here losing hydrogen atoms is oxidation), while the oxygen is getting reduced, (or gaining hydrogen atoms) and ending up as water. This reaction will take place rapidly with no further help from catalysts.
(Note that it is not a hydrogen ION (proton, H+) that is being moved about here, but the hydrogen ATOM with its electron. Actually, it is the electrons that accompany the hydrogen atoms that are fundamental to the definition of oxidation/reduction, rather than the hydrogen atoms as a whole, as we shall see later. That is, oxidation is the loss of electrons, and reduction is the gain of electrons, with or without an accompanying hydrogen ion.)
[Add another cys and -S-S- bond in folding diagram above]
The net result is tertiary structure, or the overall 3-dimensional shape of a folded-up single polypeptide. Note that there will be many regions of secondary structure within this overall tertiary structure. It is the interactions of the side chains that are to a large extent responsible for preventing the whole polypeptide from simply becoming 100% alpha-helix or 100% beta-sheet. (see picture of the tertiary structure of proteins represented by a folded ribbon, a space-filling model, a charged surface model, and by simply tracing the backbone).
(Show wire model ball with alpha-helices and beta-sheets within it. )
To review tertiary structure, try problem 2-3 part D.
So now we can see that one polypeptide molecule can be folded into a compact structure and we can understand what holds it together, but why is it that there is only one structure formed and not many? Is there only one solution to the folding problem for a particular polypeptide chain?
Perhaps all possible conformations are tried in the course of folding, and only the most stable one accumulates. Can we predict the conformation from first principles? If we plug in the properties of all the amino acid side chains, how hydrophobic they are, what is the strength of an ionic bond, etc. we can ask a computer to try and try many combinations, many interactions. This is a very difficult computer problem, even for today's supercomputers, because the number of possibilities for a good-size polypeptide of say 500 amino acids is enormous (20^500). But it has been tried, and so far usually the wrong structure comes out. The right structure is determined by examining crystals of the proteins, beaming X-rays through the protein crystals and calculating how they are refracted by the atoms in the crystal. Perhaps we really don't know the right properties of the side chains. Or perhaps there is some guide to folding that being imposed on the polypeptide as it is being polymerized in the cell, some outside influence, even a template of sorts, one can imagine a plaster mold analogy, for example. That is not the case, as we shall see next.
© Copyright 2006 Lawrence Chasin and Deborah Mowshowitz Department of Biological Sciences Columbia University New York, NY
{kind=link}
![[Purves6ed 3.4]](../purves6/figure03-04.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}