
Research Interests
Effect of Force on Protein Structural and Dynamical Properties
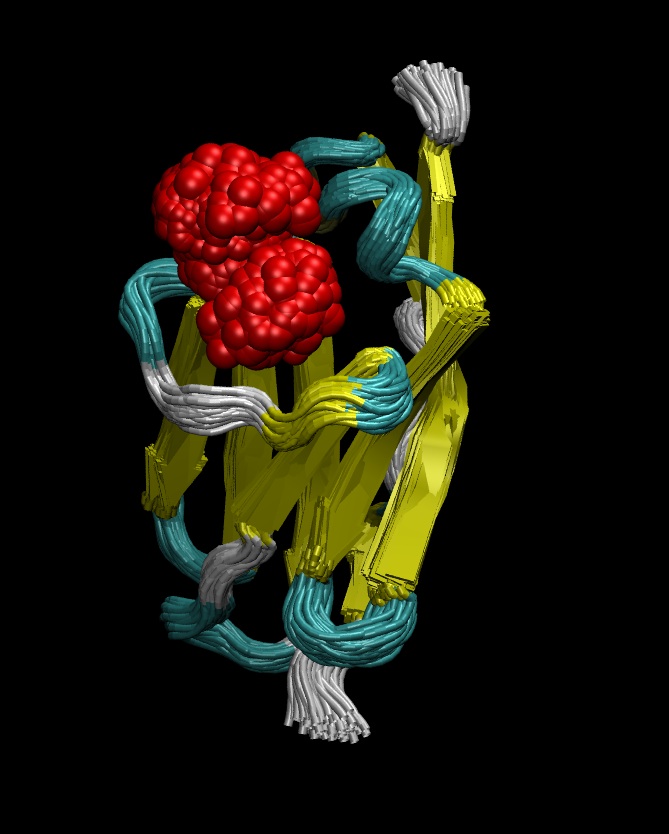
Single molecule force-spectroscopy techniques represent a whole new and exciting field providing unprecedented details about biochemical and biological mechanisms at a molecular scale. However, interpretation of the experimental data is often challenging and benefits from the perspective brought by steered molecular dynamics (SMD) simulations. An ongoing collaboration with the group of Prof. Julio Fernandez in the Biology Department has already led to significant advances in various topics, such as the effect of hydrophobic interactions in protein collaspe (1), the force-induced mechanism of protein unfolding (2) or the diffusion dynamics of proteins under force (3). In all cases, SMD simulations have been key to provide molecular details about the associated mechanisms, to help testing different hypotheses and even sometimes to predict experimental results. Current work includes the structural and dynamical aspects of protein relaxation under force or the mechanical stability of the hypothetical molten-globule states, among many other questions.
- K. A. Walther, F. Gräter, L. Dougan, C. L. Balilla, B. J. Berne, and J. M. Fernandez, Signatures of Hydrophobic Collapse in Extended Proteins Captured with Force Spectroscopy. Proc. Natl. Acad. Sci. USA, 104, 7916-7921 (2007).
- J. Li, J. M. Fernandez, and B. J. Berne, Water's role in the force induced unfolding of ubiquitin. Proc. Natl. Acad. Sci. USA, 107, 19284-19289 (2010).
- R. Berkovich, R. I. Hermans, I. Popa, G. Stirnemann, S. Garcia-Manyes, B. J. Berne and J. M. Fernandez, Rate Limit of Protein Elastic Response is Tether Dependent. Proc. Natl. Acad. Sci. USA, 109, 14416-14421 (2012).
Hydrophobicity at the Nanoscale
It has long been appreciated that water plays an essential role in determining the behavior of biological systems. The hydrophobic effect, that is the nature of solvation at oily surfaces and the assembly of such surfaces in aqueous solution, plays a crucial role in protein folding and aggregation. Our group has a long standing interest in this phenomenon, including the theory and simulation of the dewetting transiton between model hydrophobic surfaces and the study of the role of the hydrophobic effect in protiens and other biological systems. This work has uncovered the length scale dependence of the drying transition as well as the role of hydrophobic interactions in the formation of melittin tetramers. Most recently, we have studied the role of hydrodynamic interactions in the kinetics of hydrophobic collapse, and have found that such effects are essential to describing the kinetic pathway of assembly.
Below, we show two examples of hydrophobic assembly, the association of model nano-spheres and peptide aggregates. Both are driven by the large solvent fluctuations that are present as the inter-solute region dries.
- A. Wallqvist and B. J. Berne, Computer Simulation of Hydrophobic Hydration Forces on Stacked Plates at Short Range. J. Phys. Chem., 99, 2893-2899 (1995).
- X. Huang, C. J. Margulis, and B. J. Berne, Dewetting-induced collapse of hydrophobic particles. Proc. Natl. Acad. Sci. USA, 100, 11953-11958 (2003).
- P. Liu, X. Huang, R. Zhou, and B. J. Berne, Observation of a dewetting transition in the melittin tetrameter. Nature, 437, 159-162 (2005).
- R. Zangi, M. Hagen, and B. J. Berne. Effect of ions on the hydrophobic interaction between two plates. J. Am. Chem. Soc. 129, 4678-4686 (2007).
- M. G. Krone, L. Hua, P. Soto, R. Zhou, B. J. Berne, and J. Shea. Role of water mediating the assembly of Alzheimer amoyloid-beta ab 16-22 protofilaments. J. Am. Chem. Soc., 130, 11066-11072 (2008).
- J. A. Morrone, J. Li, and B. J. Berne, Interplay between hydrodynamics and the free energy surface in the assembly of nanoscale hydrophobes. J. Phys. Chem. B, 116, 378-389 (2012).
Algorithmic Development
 The development of novel algorithms and techniques is at the heart of our
work. Discoveries depend on the development of both new ways of probing
systems and methods that increase the computational effiency of
molecular simulation software. Our group has
pioneered numerous areas of methods development, including multiple time scale molecular dynamics algorithms,
techniques for path integral molecular dynamics, and the
development of polarizable model potentials and we continue to advance
these areas. Recently, we have developed novel techniques for the enchanced
sampling of biomolecular systems and pioneered tools
that account for the role of water molecules as a driving force of
ligand-protein binding. Our techniques have been widely
adopted and are implemented in many software packages.
The development of novel algorithms and techniques is at the heart of our
work. Discoveries depend on the development of both new ways of probing
systems and methods that increase the computational effiency of
molecular simulation software. Our group has
pioneered numerous areas of methods development, including multiple time scale molecular dynamics algorithms,
techniques for path integral molecular dynamics, and the
development of polarizable model potentials and we continue to advance
these areas. Recently, we have developed novel techniques for the enchanced
sampling of biomolecular systems and pioneered tools
that account for the role of water molecules as a driving force of
ligand-protein binding. Our techniques have been widely
adopted and are implemented in many software packages.
- B. J. Berne and D. Thirumalai, On the simulation of quantum systems: path integral methods. Ann. Rev. Phys. Chem., 37, 401-424 (1986).
- M. E. Tuckerman, B. J. Berne, and G. J. Martyna, Reversible multiple time scale molecular dynamics. J. Chem. Phys., 97, 1990-2001 (1992).
- S. W. Rick, S. Stuart, and B. J. Berne, Dynamical fluctuating charge force fields: Applications to liquid water. J. Chem. Phys., 101, 6141-6156 (1994).
- P. Liu, B. Kim, R. A. Friesner, and B. J. Berne, Replica exchange with solute tempering: A method of sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA, 102, 13749-13754 (2005).
- T. Young, R. Abel, B. Kim, B. J. Berne, and R. A. Friesner, Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA, 104, 808-813 (2007).
- L. Wang, B. J. Berne, and R. A. Friesner. On achieving high accuracy and reliability in the calculation of relative protein-ligand binding affinities. Proc. Natl. Acad. Sci. USA, 109, 1937-1942 (2012).
Liquids at Interfaces
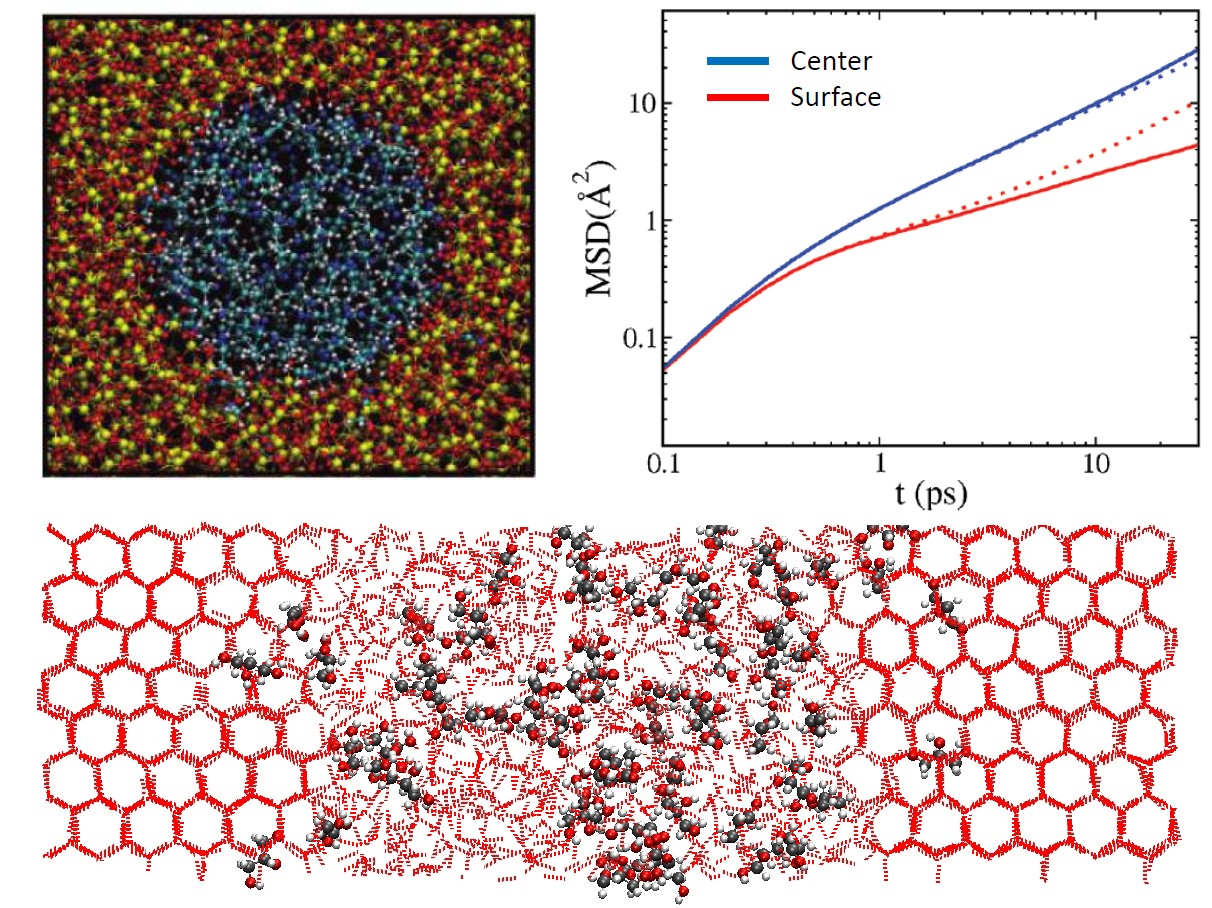 The structure and dynamics of liquids change dramatically near interfaces.
Many important physical and chemical processes are in fact defined
by the properties of the liquid-solid or liquid-air contact. Understanding
the thermodynamics of these interfaces is crucial for catalysis,
drug delivery applications, confinement in nanopores,
and understanding basic processes such as freezing.
Our group has pioneered new methods to properly quantify the behavior of
liquids at interfaces from molecular dynamics simulations.
The structure and dynamics of liquids change dramatically near interfaces.
Many important physical and chemical processes are in fact defined
by the properties of the liquid-solid or liquid-air contact. Understanding
the thermodynamics of these interfaces is crucial for catalysis,
drug delivery applications, confinement in nanopores,
and understanding basic processes such as freezing.
Our group has pioneered new methods to properly quantify the behavior of
liquids at interfaces from molecular dynamics simulations.
Initially, these new techniques were applied to the water-air interface which had important applications in atmospheric chemistry. Recently, we investigated the behavior of liquids confined in silica nanopores and showed how the dynamics of surface-hydration layer were markedly different than in the bulk center region. Our latest work investigates the antifreeze properties of small molecules which has important applications in cryobiology. Not only are these studies important in their respective fields, but they provide general insights into the complex properties of interfaces which can only be captured using fully-atomistic simulations.
- P. Liu, E. Harder, and B. J. Berne, On the Calculation of Diffusion Coefficients in Confined Fluids and Interfaces with an Application to the Liquid-Vapor Interface of Water. J. Phys. Chem. B, 108, 6595-6602 (2004).
- P. Liu, E. Harder, and B. J. Berne, Hydrogen-Bond Dynamics in the Air-Water Interface J. Phys. Chem. B, 109, 2949-2955 (2005).
- T. E. Markland and B. J. Berne, Unraveling quantum mechanical effects in water using isotopic fractionation. Proc. Natl. Acad. Sci. USA, 109, 7988-7991 (2012).
- L. Cheng, J. A. Morrone, and B. J. Berne, Structure and dynamics of acetonitrile confined in a silica nanopore. J. Phys. Chem. C, 116, 9582-9593 (2012).
Effect of Osmolytes on Protein Stability
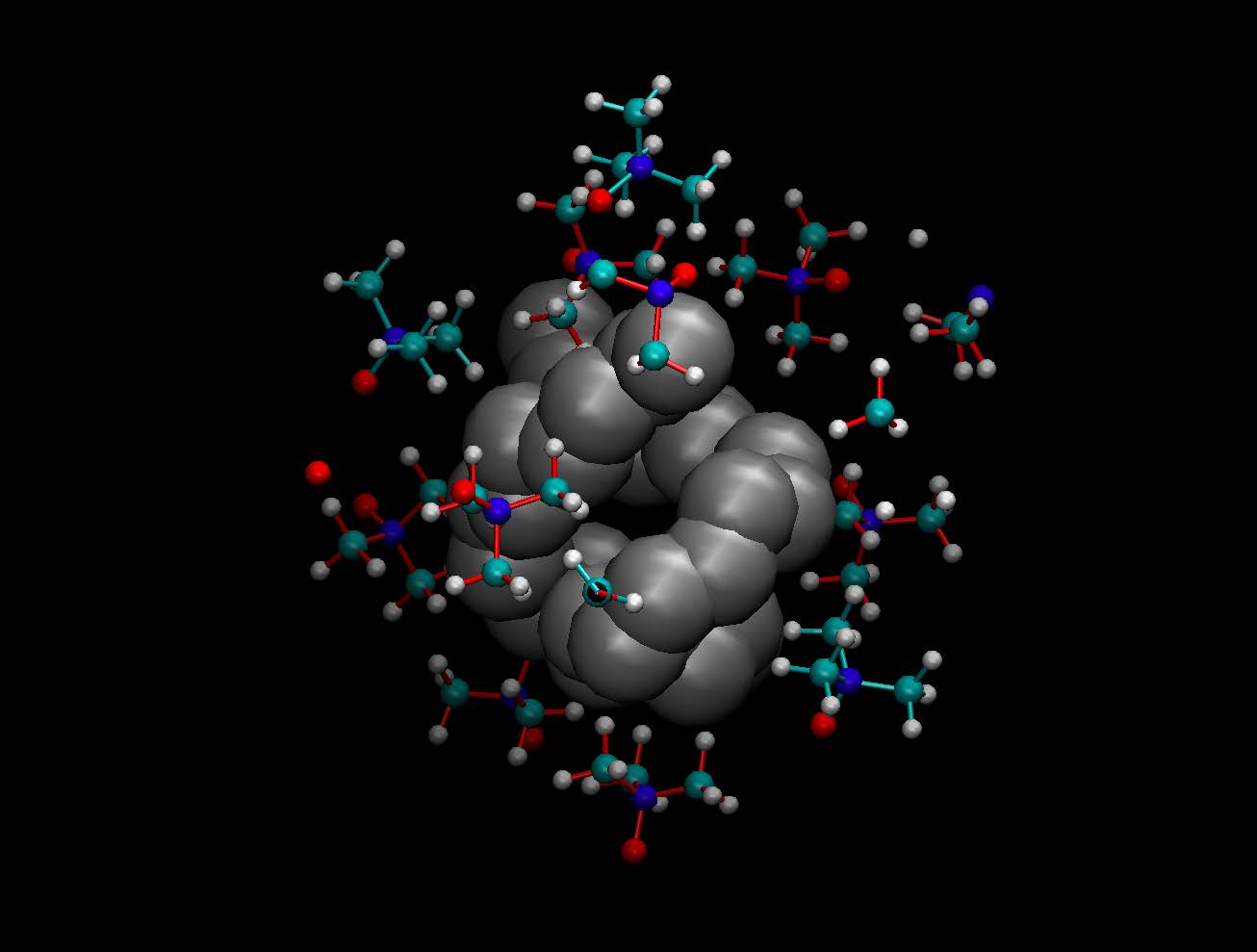
Osmolytes are small co-solvents found endogenously to protect cells against osmotic stress. However, they can have profound effects on protein stability. While some of them are denaturant (e.g. urea), others like trimethylamine N-oxide (TMAO) act as protecting osmolytes in vivo: in denaturing conditions, they bias the protein structure toward the folded state. A longstanding question has been whether their denaturing or stabilizing character arises from direct interaction between the osmolyte molecules and the protein, or instead through an indirect mechanism mediated by water molecules. Recent effort in the group has been devoted to the mechanism of urea-induced denaturation, both on model polymer systems (1) and on a real, large protein system in explicit solvent (2), a computational breakthrough at that time. These studies have shed light on how urea influences hydrophobic interactions, which are key for protein stability. They have also evidenced how urea molecules penetrates directly into the protein, subsequently exposing hydrophobic groups and later leading to full unfolding. Motivated by these successes, the molecular mechanism of protein stabilization by TMAO is now currently under investigation using similar systems and protocols.
- R. Zangi, R. Zhou, and B. J. Berne, Urea's action on hydrophobic interactions. J. Am. Chem. Soc., 131, 1535-1541, (2009).
- L. Hua, R. Zhou, D. Thirumalai, and B. J. Berne, Urea Denaturation by stronger dispersion interactions with proteins than water implies a two stage unfolding. Proc. Natl. Acad. Sci. USA, 105, 16928-16933, (2008).