Abstract
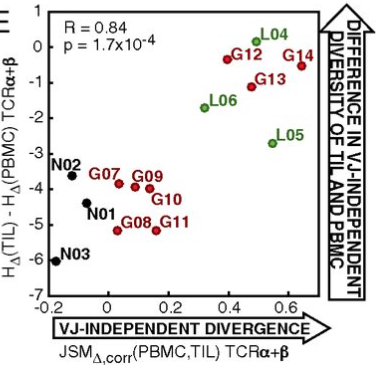
Although immune signaling has emerged as a defining feature of the glioma microenvironment, how the underlying structure of the glioma-infiltrating T-cell population differs from that of the blood from which it originates has been difficult to measure directly in patients. High-throughput sequencing of T-cell receptor (TCR) repertoires (TCRseq) provides a population-wide statistical description of how T cells respond to disease. We have defined immunophenotypes of whole repertoires based on TCRseq of the α- and β-chains from glioma tissue, nonneoplastic brain tissue, and peripheral blood from patients. Using information theory, we partitioned the diversity of these TCR repertoires into that from the distribution of VJ cassette combinations and diversity due to VJ-independent factors, such as selection due to antigen binding. Tumor-infiltrating lymphocytes (TILs) possessed higher VJ-independent diversity than nonneoplastic tissue, stratifying patients according to tumor grade. We found that the VJ-independent components of tumor-associated repertoires diverge more from their corresponding peripheral repertoires than T-cell populations in nonneoplastic brain tissue, particularly for low-grade gliomas. Finally, we identified a “signature” set of TCRs whose use in peripheral blood is associated with patients exhibiting low TIL divergence and is depleted in patients with highly divergent TIL repertoires. This signature is detectable in peripheral blood, and therefore accessible noninvasively. We anticipate that these immunophenotypes will be foundational to monitoring and predicting response to antiglioma vaccines and immunotherapy.