Abstract
BACKGROUND: Pulmonary arterial hypertension (PAH) is a rare disease characterized by distinctive changes in pulmonary arterioles that lead to progressive pulmonary arterial pressures, right-sided heart failure, and a high mortality rate. Up to 30% of adult and 75% of pediatric PAH cases are associated with congenital heart disease (PAH-CHD), and the underlying etiology is largely unknown. There are no known major risk genes for PAH-CHD.
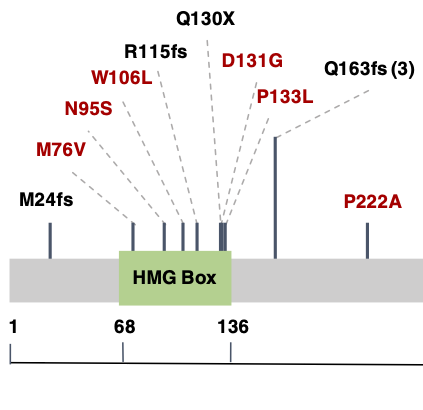
METHODS: To identify novel genetic causes of PAH-CHD, we performed whole exome sequencing in 256 PAH-CHD patients. We performed a case-control gene-based association test of rare deleterious variants using 7509 gnomAD whole genome sequencing population controls. We then screened a separate cohort of 413 idiopathic and familial PAH patients without CHD for rare deleterious variants in the top association gene. RESULTS: We identified SOX17 as a novel candidate risk gene (p = 5.5e-7). SOX17 is highly constrained and encodes a transcription factor involved in Wnt/β-catenin and Notch signaling during development. We estimate that rare deleterious variants contribute to approximately 3.2% of PAH-CHD cases. The coding variants identified include likely gene-disrupting (LGD) and deleterious missense, with most of the missense variants occurring in a highly conserved HMG-box protein domain. We further observed an enrichment of rare deleterious variants in putative targets of SOX17, many of which are highly expressed in developing heart and pulmonary vasculature. In the cohort of PAH without CHD, rare deleterious variants of SOX17 were observed in 0.7% of cases.
CONCLUSIONS: These data strongly implicate SOX17 as a new risk gene contributing to PAH-CHD as well as idiopathic/familial PAH. Replication in other PAH cohorts and further characterization of the clinical phenotype will be important to confirm the precise role of SOX17 and better estimate the contribution of genes regulated by SOX17.