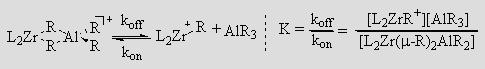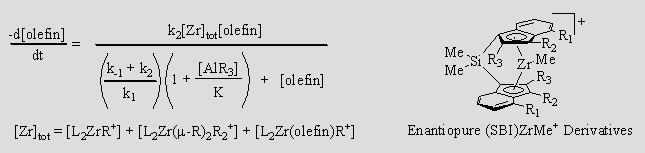|
||
|
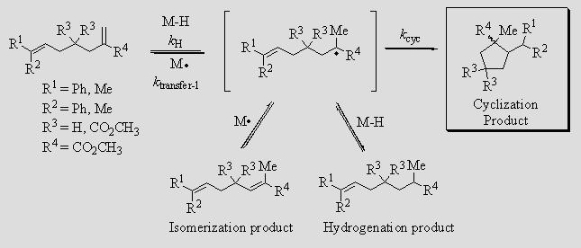
Tin- Free and Catalytic Radical Cyclizations Using Hydrogen Atom Transfer from Transition-Metal Hydrides The tin hydrides now used stoichiometrically for radical cyclization in the laboratory are too toxic, and too hard to remove, for use in the manufacture of pharmaceuticals. Furthermore, tin hydrides require that some other heavy element, typically iodine, bromine, selenium, or sulfur be used stoichiometrically to generate the initial radical. Our research is focused on the development of transition-metal hydrides as a non-toxic, tin-free, radical cyclization methodology.We have determined the rate constants kH for H∙ transfer from CpCr(CO)3H to olefins with various substitution patterns from the observed rates of H/D isotope exchange, hydrogenation, or both. We have used these results to design substrates that can be induced to undergo radical cyclization by H∙ transfer.
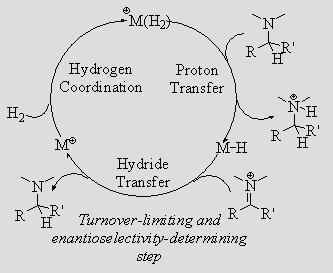
With CpCr(CO)3H, the reaction is both tin-free and catalytic, as CpCr(CO)3∙ is converted back to the hydride under an H2 atmosphere. With various vanadium hydrides, HV(CO)4P-P (P-P = dppb, dppp, dppe, dppm), the reaction is faster but stoichiometric. The V-H bond in these vanadium hydrides is appreciably weaker than the Cr-H bond (54.9-57.9 kcal/mol vs. 61.5 kcal/mol). Presumably the V-H bonds are so weak as to make the reaction of V∙ with H2 unfavorable. Other metal-hydride complexes, especially those with increased steric bulk, will be investigated as this should improve the ratio of cyclization to hydrogenation/isomerization. Additional research in this area includes investigating other substrate possibilities for cyclization. Questions to be answered include: How slow a kcyc can our H∙-transfer-initiated cyclizations tolerate? At what rate do triple bonds receive H∙? Can we obtain a cyclized product with a vinyl end-group through H∙ abstraction from the cyclized radical by M∙? Such a reaction would be catalytic without hydrogen pressure. Ionic Hydrogenation Hydrogenation by an ionic mechanism, involving H– and H+ transfer, is often selective for polar bonds (C=N and C=O). We have demonstrated that half-sandwich Ru(II) hydride complexes are effective catalysts for the ionic hydrogenation of iminium and aziridinium cations. We are designing and preparing catalysts that will perform these reductions with high enantioselectivities.
Although ionic hydrogenation normally delivers H2 as H– and H+, we are also interested in delivering H2 as e–, H•, and H+. Such a mechanism should allow the regioselective hydrogenation of epoxides to primary alcohols. Currently, no catalyst is capable of performing this transformation with H2.
As this reaction requires the initial transfer of an electron from the catalyst, we are interested in the electrochemical properties of electron-rich metal hydride complexes.
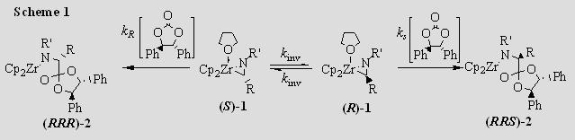
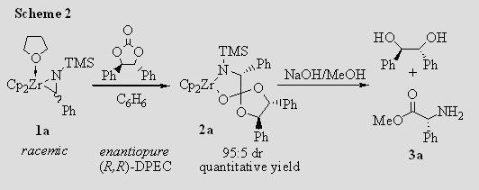
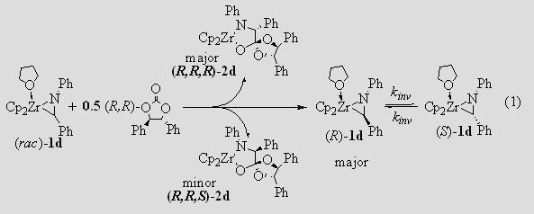
Insertion Chemistry of Metallaziridines We have reported the synthesis of amino acid esters and a-amino amidines from the insertion of cyclic carbonates and carbodiimides into the Zr–C bonds of zirconaaziridines. The insertion of a chiral cyclic carbonate can effect a dynamic kinetic asymmetric transformation (Scheme 1) if the insertion is slower than the interconversion of the two zirconaaziridine enantiomers. If the chiral reagent is added slowly, so that kR[carbonate] and kS[carbonate] are smaller than kinv, one of the diastereomeric insertion products is produced in excess, leading to an enantioenriched amino acid ester (Scheme 2).
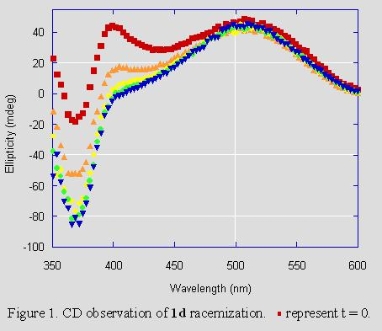
We need to know kinv (the rate constant for enantiomer interconversion) in order to determine how slowly the chiral carbonate must be added for maximum diastereoselectivity. We have measured kinv for a variety of zirconaaziridines by using CD spectroscopy to monitor the racemization of an unequal mixture of the enantiomers of 1 (Figure 1). We generate that unequal mixture by adding 0.5 equiv of the chiral carbonate quickly to the racemic zirconaaziridine (eq 1).
The results agree well with laboratory experience. The values of kinv we measure agree with the rates of addition we find necessary. We are investigating the synthesis and reactivity of other Group IV metallaaziridines and the use of other chiral insertion reagents.
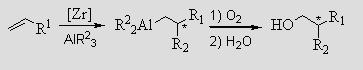
Asymmetric Carboalumination of a-Olefins and Ethylene Oligomerization Zr/Al transmetallations are vital in a) zirconium catalyzed carboalumination of olefins and b) the oligomerization of olefins such as ethylene.
Carboalumination is useful as a route to chiral primary alcohols.
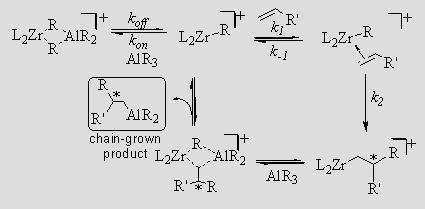
Zr/Al transmetallations play an important role in the industrially relevant oligomerization reactions of olefins such as ethylene. Addition of excess AlMe3 yields oligomers with narrow molecular weight distributions. Understanding how transmetallation is affected by factors such as chain length, chain branching, and chain type is vital to producing polymers with a precise molecular weight distribution, or a precise architecture as a result of chain-shuttling polymerization. In solution, catalytically active zirconium alkyl cations form heterobimetallic species with trialkylaluminums. Such heterobimetallics appear to be responsible for transmetallation. We have measured the dissociation rate constant, koff, for a variety of such heterobimetallic species and have shown that it predicts carboalumination activity.
This finding is relevant to both ethylene oligomerization and carboalumination because each involves the insertion of olefin into the Zr–R bond of L2ZrR+, and subsequent transmetallation from Zr to Al. If kon[AlR3] and koff are fast enough, L2ZrR+ and L2ZrCH2CHR'R+ will be in equilibrium with their respective AlR3 adducts, and chains will be transmetallated onto Al as fast as they grow on Zr. The rate of olefin uptake will depend on how much L2ZrR+ and L2ZrCH2CHR'R+ remain uncomplexed by AlR3.
The value of K seems, not unreasonably, to depend on koff, so catalysts with larger values of koff should be more effective catalysts. This prediction appears to be true and we have observed that (SBI)ZrR+ has a relatively large koff and is an effective catalyst for carboalumination. We are thus preparing, resolving, and testing enantiopure derivatives of (SBI)ZrR+ as catalyst for asymmetric carboalumination. If our mechanism is correct the rate law should be the one below, which we are testing.
We are conducting kinetic experiments that probe how transmetallation varies with different chain sizes, types and branching. We are also investigating the detailed kinetics of chain growth on aluminum. |
||