Softwares
Sigma-P Method for Rare-Variant Analysis
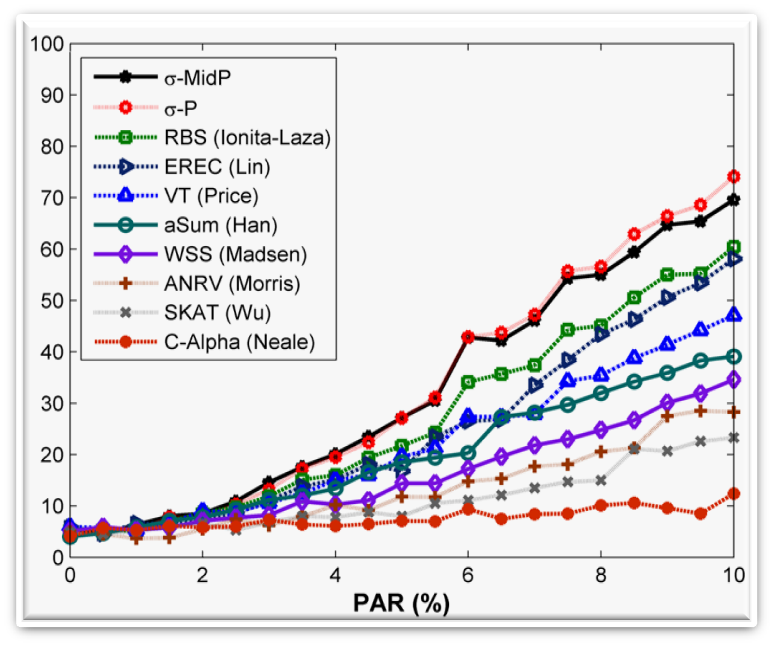 |
SigmaP is a rare-variant method for detecting disease associations in case-control sequencing studies. The Sigma-P statistic aggregates the effects of multiple variant sites by computing a weighted sum of the log p-values per site. Each site is weighted by the inverse of its expected standard deviation (denoted by sigma) of the number of variants in controls. The method is robust against signal noise introduced by a large number of neutral variants and is effective for handling variants with opposite effects. Download: R Scripts and Sample Data |
Penalized Conditional/Unconditional Logistic Regression - pclogit R package
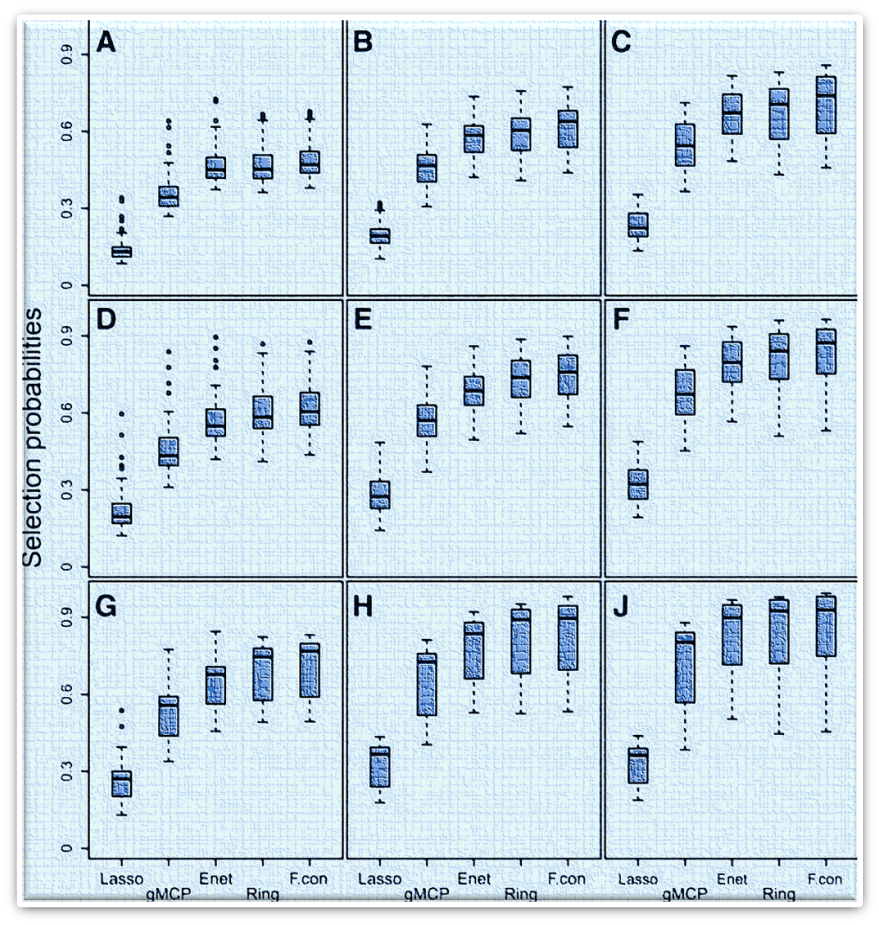 |
pclogit is an R package for penalized conditional/unconditional logistic regression using a network-based peanlty for matched/unmatched case-control data with grouped or graph-constrained variables. The algorithm is efficient for fitting the regularization path and for providing selection probabilities of each predictor for the anaylsis of high-dimensional matched/unmatched case-control data. It uses cyclical coordinate descent in a pathwise fashion. Downloads: Manual, pclogit.tar.gz (for Linux/Unix only) |
Rare variants selection - rvsel R package
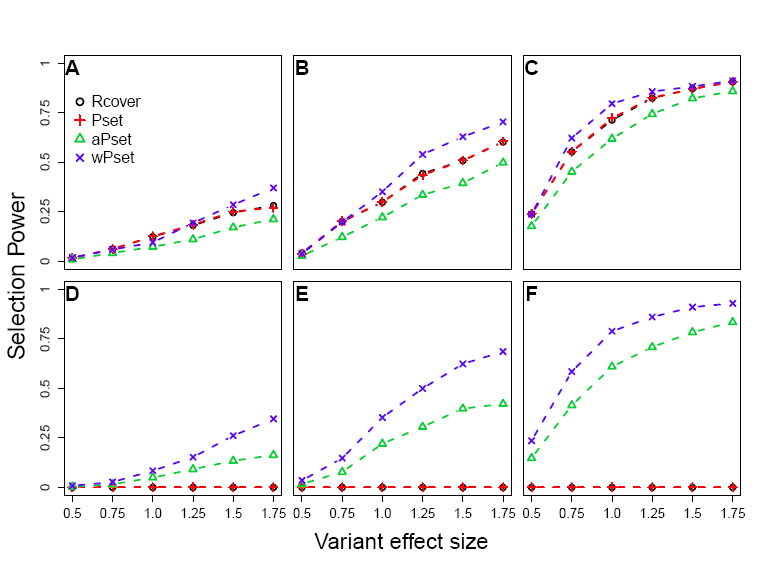 |
rvsel is an R package for rare variants selection with sequence data. The most outome-related rare variants are selected within a gene or a genetic region. The selection procedure is based on the power set of the subset of the rare variants. Downloads: Manual, rvsel_0.1.tar.gz (for Linux/Unix only) |
A Network-assisted algorithm for Epigenetic studies - NEpiC R package
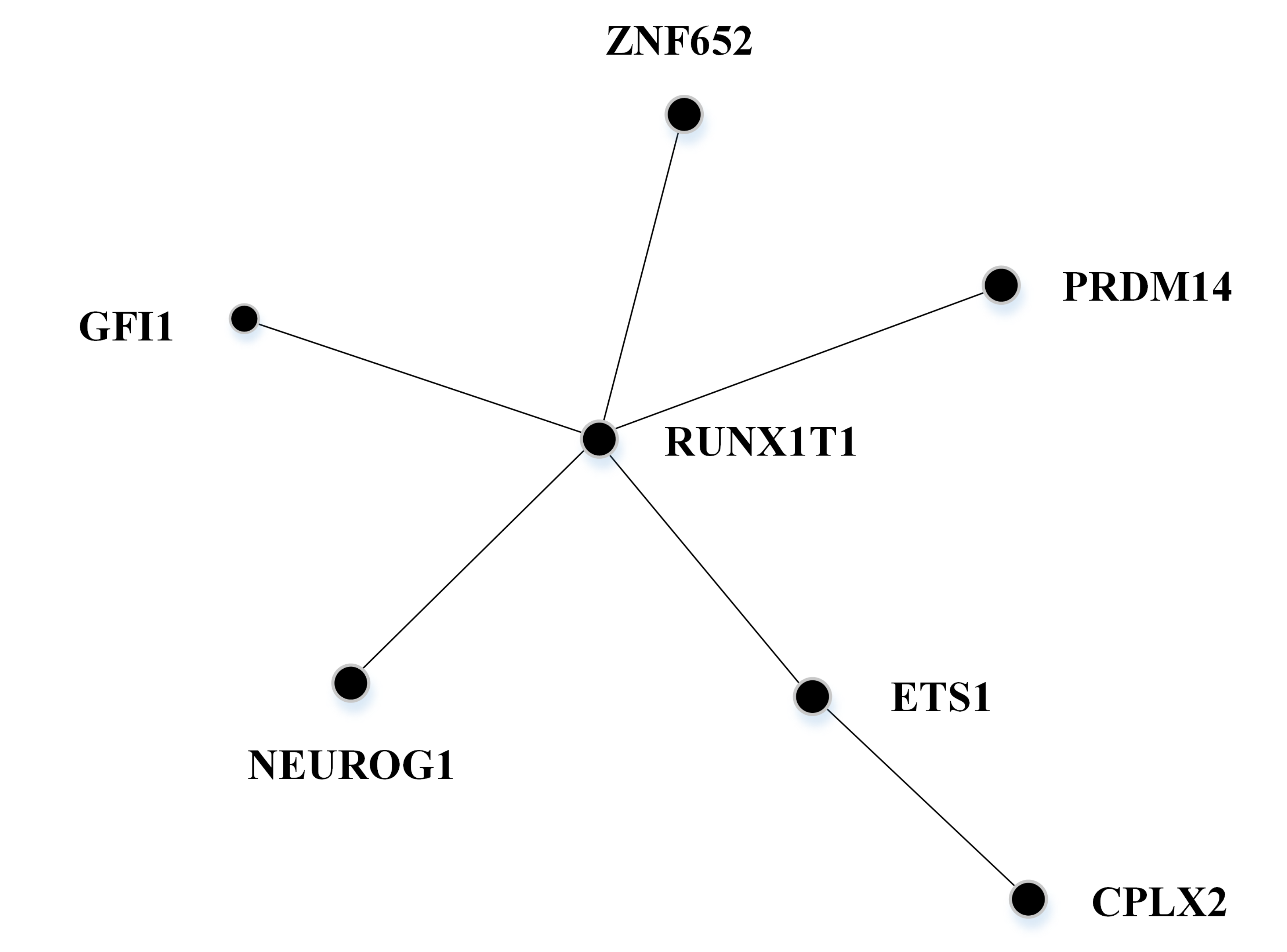 |
We present a network-assisted algorithm, NEpiC, that combines both mean and variance signals in searching for differentially methylated sub-networks using the protein-protein interaction (PPI) network. Download: R Scripts and Sample Data |
A penalized Exponential Tilt Model for Epigenetic studies - pETM R package
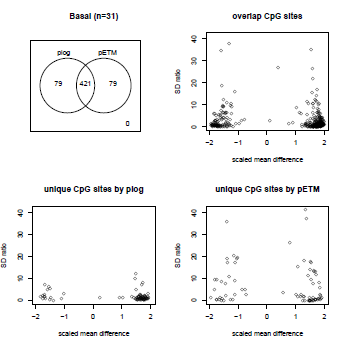 |
We present a penalized Exponential Tilt Model (pETM) using network-based regularization that captures both mean and variance signals in DNA methylation data and takes into account the correlations among nearby CpG sites. Download: R package and manual |
The MiAge Calculator
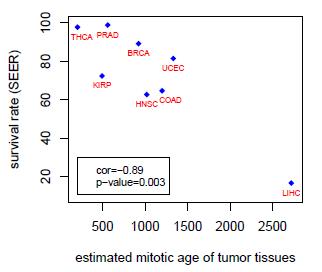 |
To estimate the number of cell divisions (mitotic age) of a given tissue type between individuals is of great interest as that allows their stratification of prospective cancer risk. Here we introduce the MiAge Calculator, a DNA methylation-based mitotic clock calculator based on a novel statistical method MiAge, designed to quantitatively estimate mitotic age of a tissue of an individual. This R code is for the new DMR detection algorithm we proposed that uses mean and variance combined signals. Download: R Scripts and Sample Data |
The Pan-Cancer Analysis
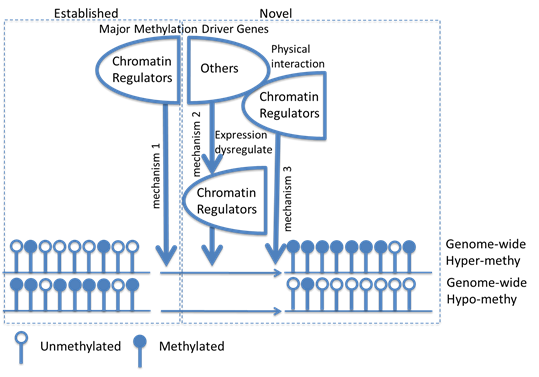 |
MutIng algorithm integrates somatic mutation and DNA methylation data of multiple cancers and identifies methylation driver genes (MDGs) that, when mutated, have strong associations with specific methylation changes across cancer types. Download: R Scripts and Sample Data |
Differentially Methylated Regions (DMRs) detection
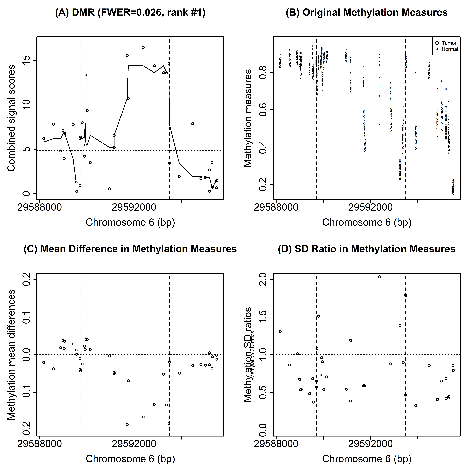 |
Most existing methods developed to identify differentially methylated loci (DML) use mean signals only, and only a few methods were developed to identify DML using both mean and variance signals, while all existing methods to detect differentially methylated regions (DMRs) focus on mean signals only. This R code is for the new DMR detection algorithm we proposed that uses mean and variance combined signals. Download: R Scripts and Sample Data |
The Epigenetic-Distance Method
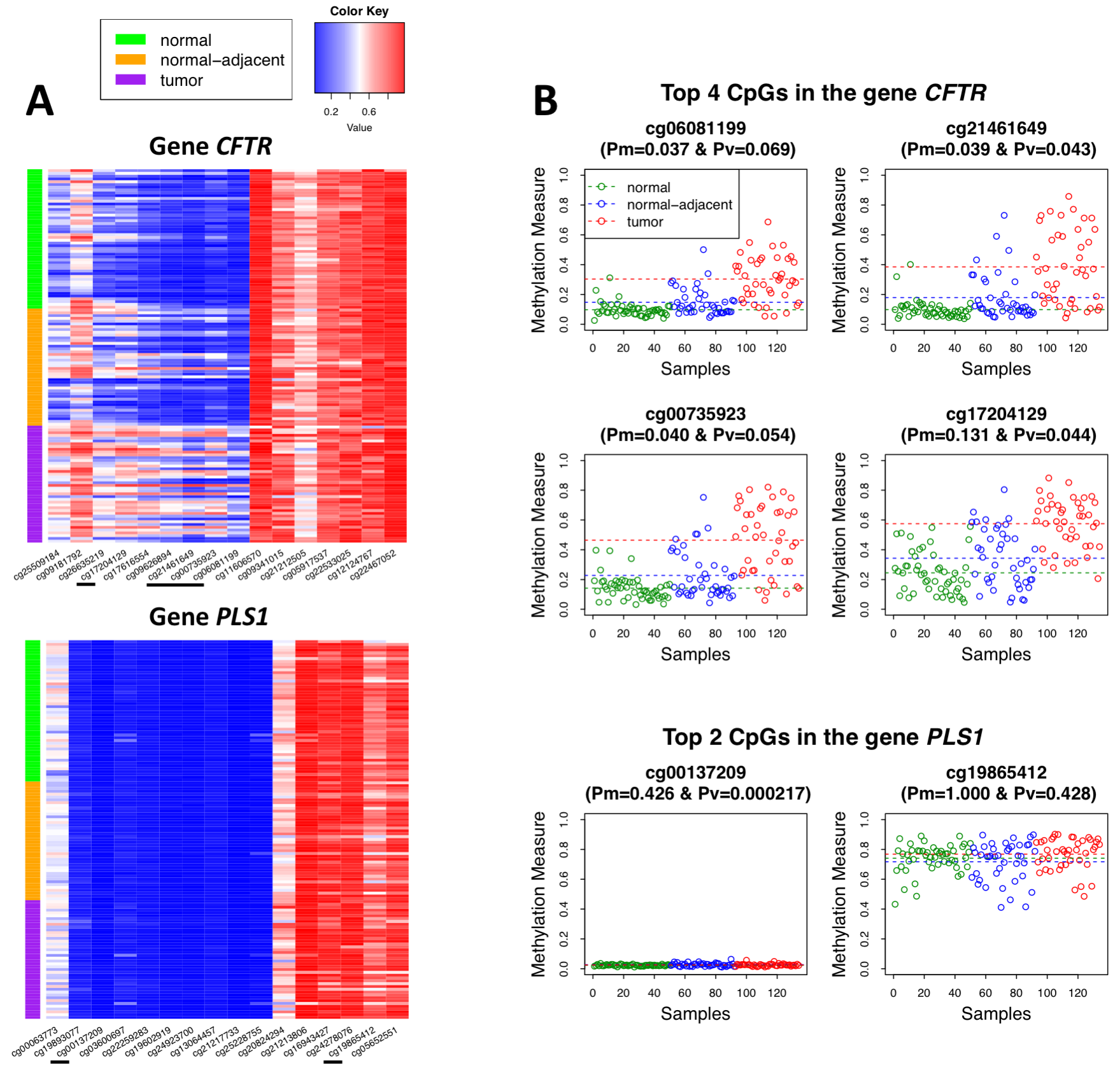 |
We developed a weighted epigenetic distance-based method characterizing (dis)similarity in methylation measures at multiple CpGs in a gene or a genetic region between pairwise samples, with weights to up-weight signal CpGs and down-weight noise CpGs. Using distance-based approaches, weak signals that might be filtered out in a CpG site-level analysis could be accumulated and therefore boost the overall study power. In constructing epigenetic distances, we considered both differential methylation (DM) and differential variation (DV) signals. Download: R Scripts and Sample Data |
ab-SNF: the association-signal-annotation boosted SNF (Similarity Network Fusion) Method
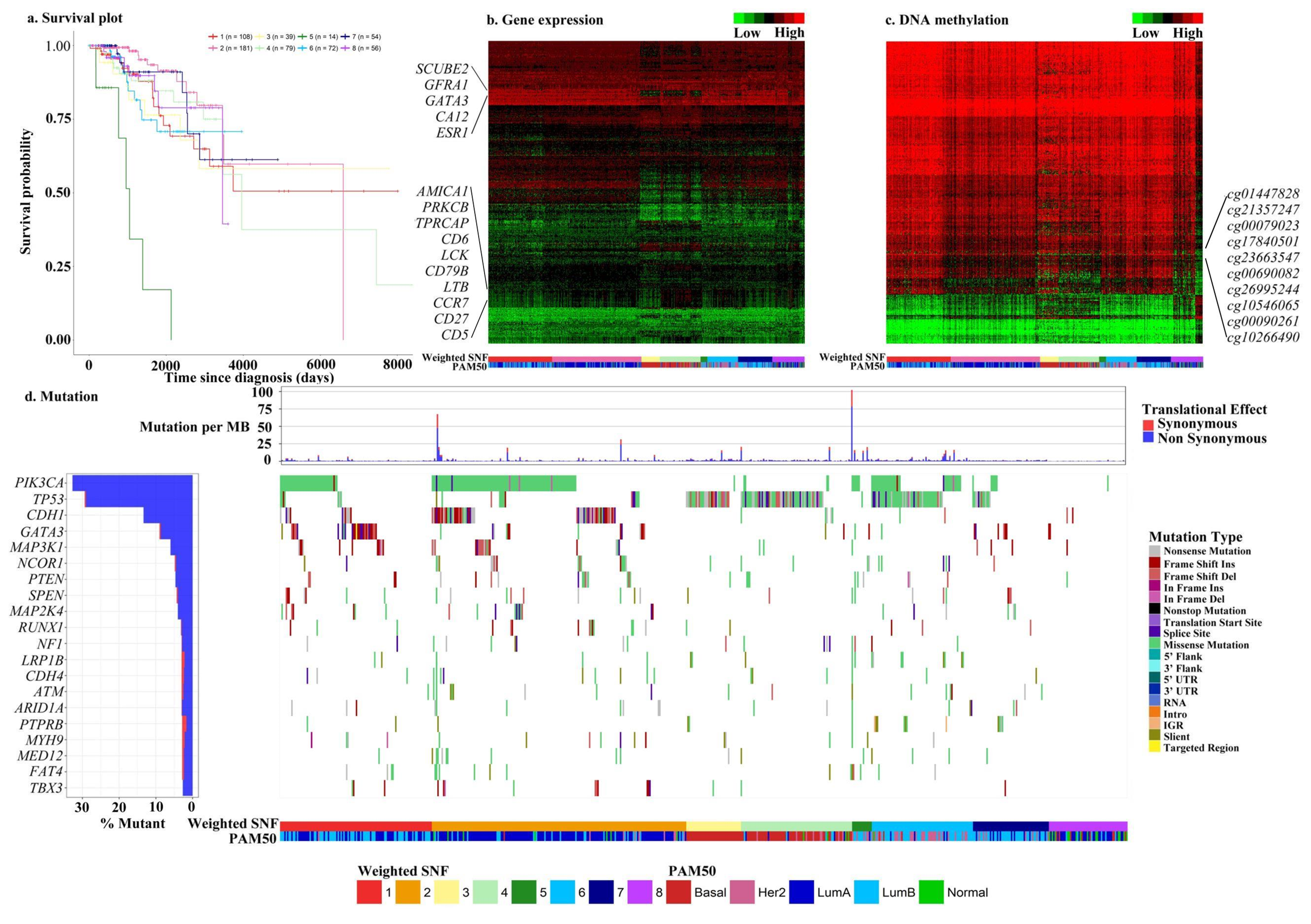 |
The association-signal-annotation boosted similarity network fusion (ab-SNF) method adds feature-level association signal annotations as weights when constructing pairwise similarity measures between subjects aiming to up-weight signal features and down-weight noise features to improve the performance in disease subtyping. Download: R Scripts and Sample Data |
Dw-main-int: method for Interactions between DNA methylation and environmental factors
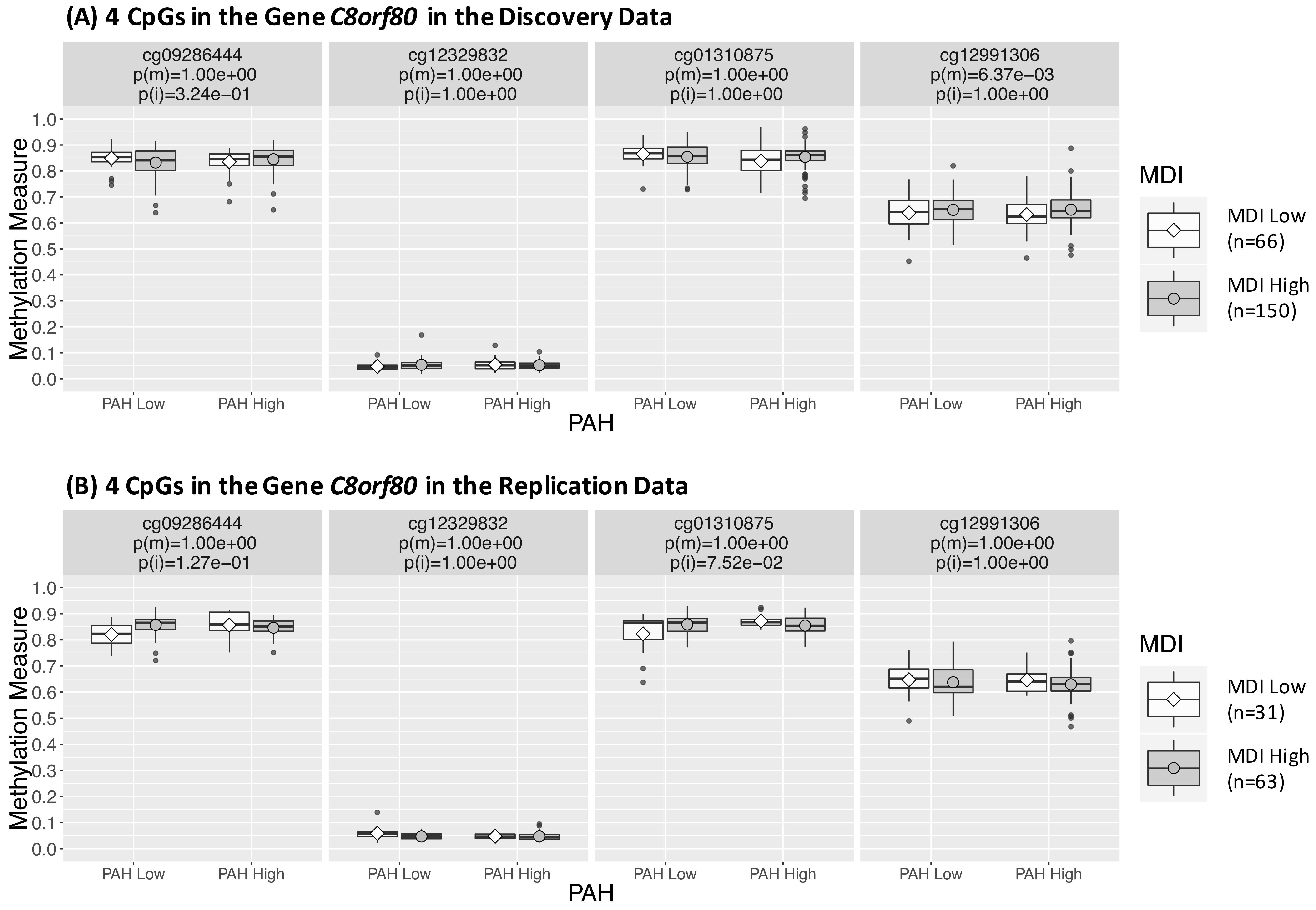 |
We developed a weighted epigenetic distance-based method with a pseudo-data matrix constructed with cross-product terms between DNA methylation and environmental factors that are able to capture their interactions on health outcomes. The distances between pairs of subjects can then be calculated combining the original data matrix with measures of DNA methylations and environmental factors together with the pseudo-data matrix with interactions. Using this approach, we can identify both main and interaction effects. Download: R Scripts and Sample Data |
DiSNEP: a Disease-Specific gene Network Enhancement to improve Prioritizing candidate disease genes
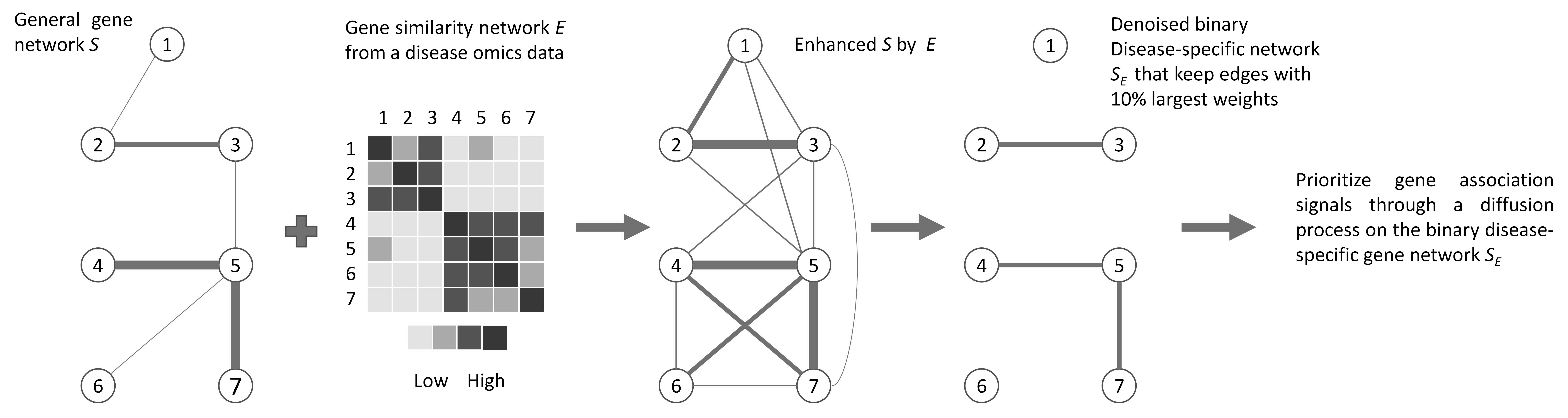 |
We developed a framework, DiSNEP, that enhances a general human gene network into a network for a specific disease that better reflects true gene interactions for the disease, which subsequently improves network-assisted candidate gene prioritization. https://github.com/pfruan/DiSNEP" |