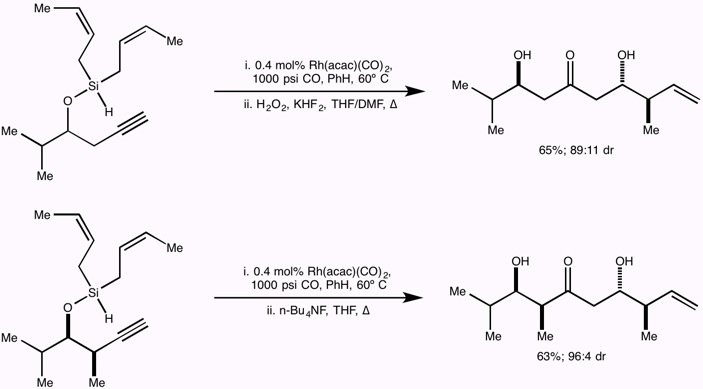
Research Summary
Research in the Leighton group is focused on the development of stereoselective reactions with relevance to both medicinal and natural products chemistry, the development of new strategies for the efficient synthesis of polyketide natural products with a particular emphasis on tandem reactions, the application of these methods to the total synthesis of polyketide natural and unnatural products, and finally on the total synthesis of architecturally complex polycyclic natural products of biological significance.
The guiding philosophy for all of these programs is that the methods we develop should be user-friendly and experimentally trivial, and environmentally and economically sound, all while providing access to otherwise inaccessible targets of structural and/or biological significance with exquisite levels of stereocontrol.
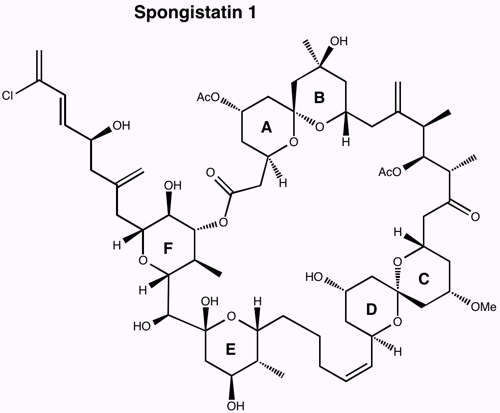
Total Synthesis of Architecturally Complex Polycyclic Natural Products
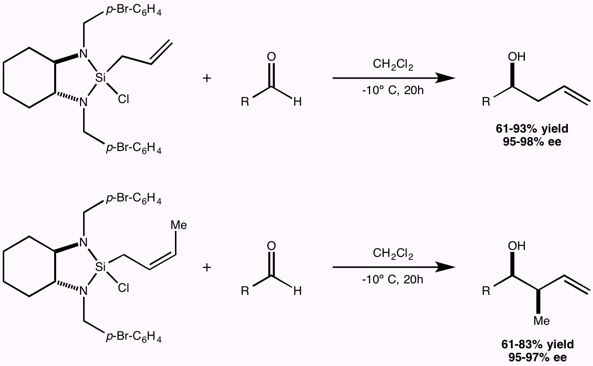
New Ligand and Catalyst Systems for Highly Practical and Enantioselective Reactions