
EXPERIMENT 3
Liquid-liquid Extraction and Recrystallization
BACKGROUND
Extractions
Extraction is one of the oldest chemical operations known; it involves transferring a solute from one phase to another. The preparation of a cup of tea or coffee represents a process of extraction of flavor and odor components from dried material into water. When a compound is extracted from a solid material into a liquid, the process is referred to as a solid-liquid extraction; if the transfer occurs from one liquid into another is called liquid-liquid extraction. Most organic synthetic procedures are followed by workups employing extraction to isolate the product of interest.
How would a mixture of saccharin and NaCl be separated? Both are soluble in water. However, saccharin is somewhat soluble in diethyl ether while salt is not. If you dissolve the mixture in water and then add ether, two layers will form because ether and water are immiscible. Most of the saccharin will be extracted into the ether layer. Notice that the separation is not absolute. The salt and saccharin (the solutes) are distributed between the two solvents, and a dynamic equilibrium is established. The ratio of the concentration of a solute in a second solvent (e.g. ether) to its concentration in water is constant, called the partition coefficient K:
Ksolute = Cether / Cwater
Cether and Cwater represent the molar concentration of the solute in ether and water respectively.
This constant depends on the solvent used, the solute itself, and temperature. In this particular case Ksaccharin is a large number because saccharin is more soluble in ether than water while Ksalt is a small number because salt is slightly soluble in ether. Using this constant, one can show that extracting a component from a mixture several times with small portions of solvent is more efficient than extracting it with one large portion.
The separatory funnel is the tool of trade for liquid-liquid extraction. In order to increase the surface area between the two layers, and speed the attainment of equilibrium, the separatory funnel is shaken and vented. The organic layer (ether) is then separated from the aqueous layer, and dried. Any water dissolved in the ether can be removed by utilizing a drying agent such as anhydrous magnesium sulfate (MgSO4) and filtering off the hydrate (MgSO4•xH2O) that forms. Another way to dry an ether layer is to wash it with saturated NaCl solution (brine) before adding drying agent. The brine transfers the water from the ether layer to the aqueous layer. The dry ether solution is evaporated by a rotary evaporator (see the Instrumentation Guide) and the solute remains in the flask. If the ether is not properly dried, the remaining solute in the flask will be moist.

Other organic solvents that are used in extractions include ethyl acetate (CH3CO2C2H5), methylene chloride (CH2Cl2), chloroform (CHCl3), hexane (CH3(CH2)4CH3), and benzene (C6H6). Benzene and chloroform are usually avoided as solvents due to their carcinogenic nature. Methanol and ethanol are not useful extraction solvents because they are miscible with water and will not form a separate layer. Chloroform and methylene chloride are denser than water, while most other organic solvents are not as dense as water. Therefore, the organic layer could be above or below the aqueous layer depending on the organic solvent used. If you are not sure which layer is the organic or the aqueous layer, perform the water drop test: add a drop of either layer on top of a watch glass filled with water. The aqueous layer will readily mix with water. You may need to add several drops, as some solvents have a small, but significant, solubility in water.
The formation of an emulsion is a common problem when performing extractions. An emulsion is a stable dispersion of one liquid in a second immiscible liquid. Emulsions delay the separation of two liquids, making it necessary to “break” the emulsion. This can be done mechanically (settlers, cyclones, centrifuges, filtration through Celite) or chemically (addition of salt or a saturated NaCl solution called brine). The addition of salt increases the surface tension of the droplets and increases the density of the aqueous layer, thereby forcing separation. If one of the solvents being used is water, the addition of a saturated aqueous sodium chloride solution will help destroy the emulsion. You should avoid shaking a solution that tends to form emulsions.
Depending on the impurities being removed extractions can be classified as :
- •Aqueous extraction
- •Acidic extraction
- •Basic extraction
Aqueous Extraction
An organic mixture is extracted with water to remove highly polar materials such as inorganic salts, strong acids or bases, and low molecular weight polar substances. Normally, water extractions are used immediately following extractions of a mixture with either an acid or base to ensure that all traces of the acid or base have been removed.
Acidic Extraction
Extracting an organic mixture with a dilute acid (5% HCl) removes any basic impurities such as amines. Bases are converted to their cationic salts by the acid.
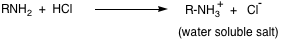
This cationic salt can be converted to its neutral form by adding base to the acid extract.
Basic Extraction
Extracting an organic mixture with a dilute base (5% sodium bicarbonate or NaOH) converts any strongly acidic impurities to their anionic salts.
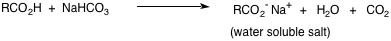

This anionic salt then can be regenerated to its neutral form by acidifying the basic extract.
Purification by Recrystallization
The technique of recrystallization is used to purify inorganic and organic compounds in the solid state. Essentially, impurities are separated from the desired product by selecting a solvent, or a solvent mixture, that will keep the impurities in solution at all temperatures, thus prohibiting these impurities from precipitating along with the product crystals. The goal is to prepare a saturated solution of solute in the solvent at its boiling point and allow it to cool. If the solvent or solvent mixture is properly chosen, the compound will have decreased solubility at lower temperatures, and the solution will precipitate crystals as it cools.
Recrystallization Using a Single Solvent
Ideally, the solute is very soluble in the solvent at its boiling point, but virtually insoluble at 0°C. Usually, recrystallization is carried out by first dissolving the solid in a boiling hot solvent. Next, the solution is cooled below room temperature, and the crystals that crash out are collected by vacuum filtration. To do this the substance to be recrystallized is placed in an Erlenmeyer flask and a minimal amount of hot solvent is added to dissolve the solid, as the solvent is heated to maintain it at its boiling point. The solution is then allowed to slowly cool to room temperature undisturbed. Allowing crystals to grow slowly is important because crystals consisting entirely of the same repeating unit will have the most uniform and strongest intermolecular interactions. For pure crystals to grow, however, there must exist a thermodynamic equilibrium between solid- and dissolved-phase solute. If the solution is shocked and cooled rapidly, crystals will grow haphazardly and are more likely to incorporate impurities.
Once recrystallization is complete, the crystals must be separated from the mother liquor via suction filtration, washed a few times with the appropriate ice-cold solvent (to discourage the now-recrystallized solid from dissolving) and dried either in air or in a desiccator.
Recrystallization Using Solvent Mixtures
In many cases, a single solvent does not fit the above requirement satisfactorily, and as a result, a mixture of solvents is used. By using a solvent mixture, we are attempting to create a solvent system that closely resembles an ideal solvent as shown in the Figure below.
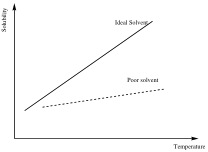
Figure 1. Ideal- and poor-recrystallization solvents.
In this situation, a solvent is chosen that will readily dissolve the solid. After dissolution, the system is filtered to remove any solid impurities (if necessary). A second solvent miscible with the first, but in which the solute has little solubility, is then added dropwise to the hot solution to achieve saturation. The first solvent in the dual solvent system readily dissolves the solid at high temperatures. However, at the boiling point of this first solvent, the solution is not saturated, and thus, this single solvent does not exhibit ideal behavior. Upon addition of the second solvent, the solution approaches saturation at the boiling point of this solvent system. This new dual solvent system best approaches ideal solvent behavior.
General Procedure
The material to be recrystallized is dissolved in the minimum amount of the solvent in which the compound is most soluble at its boiling point. While the solution boils, the solvent in which the compound is less soluble is added drop wise until it just turns the solution cloudy. If necessary, a little of the more-soluble solvent is added to clear the mixture; as the mixture cools, the desired compound will crystallize out. When using mixed solvents keep in mind that that they must be miscible so that separate layers do not form.
Table 1. Some Miscible Solvent Pairs For Recrystallization
Methanol - Water Ether - Acetone
Ethanol - Water Ether - Petroleum Ether
Ether - Methanol Methylene Chloride - Methanol
Ethyl Acetate – Hexanes Ethyl Acetate - Ether
In this list, the first solvent listed is the principal solvent used to dissolve the “impure” crystals. The second solvent, present in significantly lower quantity, enables saturation of the solution and thus initiates the gradual precipitation.
Melting Points
Once an organic solid has been isolated, the melting point range is measured to establish the compound’s identity and purity. The melting point of a solid compound is the temperature at which a phase transition from solid to liquid occurs. This is a demonstration of colligative properties, which can be rationalized by the lowering of the vapor pressure of pure liquids due to the presence of impurities.
It should be apparent that the impurity must be soluble in the compound in order to cause a melting point depression; i.e., an insoluble impurity such as sand or charcoal will not depress the melting point. The impurity does not need to be a solid. It can be a liquid such as water or an organic solvent. Melting points are generally measured and reported as a range rather than as a single discrete temperature.
EXPERIMENTAL OUTLINE
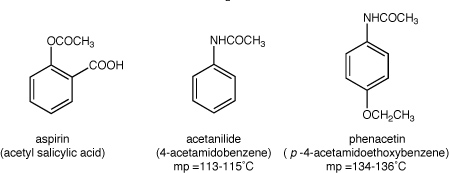
In this experiment you will separate a mixture of aspirin and an unknown that is either acetanilide or phenacetin by making use of their solubility and acid-base properties. You will purify the unknown by recrystallization and determine which of the two substances it is based on its melting point.
PROCEDURE
PART 1. Separation of Aspirin and Unknown
Before you begin, discuss how you might separate a mixture of aspirin and either acetanilide or
phenacetin with you group. How can you take advantage of the acid-base properties of the compounds and their solubilities in aqueous and organic solvents?
- 1. Weigh about 3 g of the unknown mixture and transfer to a clean, dry 125 mL Erlenmeyer flask. Dissolve in approximately 50 mL dichloromethane. Dichloromethane may be harmful if ingested, inhaled, or absorbed through the skin. Minimize contact with the liquid and handle it under the fume hood.
- 2. Prepare at least 100 mL of a saturated sodium bicarbonate solution (about 10% w/v). Add your solution from step 1 to a seperatory funnel and extract the aspirin in two 25 mL portions of NaHCO3. Your mentor will demonstrate the proper use of a seperatory funnel. Collect the organic and aqueous layers in separate Erlenmeyer flasks. Why will aspirin be extracted into the aqueous layer?
- 3. Set your organic layer aside. Slowly add 6 M HCl to your aqueous layer while stirring until the pH is about 2. Cool the solution and filter off the solid by vacuum filtration. Wash your solid with cold distilled water and dry to constant mass. Take the melting point.
- 4. Dry your organic layer with sodium sulfate and gravity filter into a pre-weighed round-bottom flask. Evaporate the solvent using a rotovap and determine the mass of solid you obtain. Determine the melting point of your crude solid.
PART 2. Recrystallization of Unknown
- 5. Dissolve your solid in a minimal amount of boiling water in an Erlenmeyer flask. Why is an Erlenmeyer flask ideal for recrystallizations?
- 6. If the solution is colored, add a small amount of activated carbon and gravity filter the hot solution into a second flask. Add additional hot solvent to your solution before filtering and use a funnel pre-heated with vapors from your boiling solvent to prevent recrystallization and loss of product.
- 7. Heat your solution until the solute is completely dissolved and then allow it to cool to room temperature. Cool for 10-15 minutes on an ice bath to complete the recrystallization and collect the crystals by vacuum filtration.
- 8. While your solution cools, convince yourself that water is an appropriate solvent for recrystallization by performing three solubility tests. Take about 10 mg of your crude unknown (the tip of a spatula) and place in a test tube with about 0.3 mL of either distilled water, hexanes, ethyl acetate, acetone, or ethanol. Observe the degree to which the solid dissolves at room temperature, at 0°C, and at the solvent’s boiling point.
- 9. After your recrystallized unknown is sufficiently dry, determine its melting point and identify it as either acetanilide or phenacetin. Confirm your results by grinding a 50/50 mixture of your unknown and a pure sample of the compound you suspect, and determine the melting point. How does this technique work to confirm your result?