C–C Bond Cleavage
The cleavage of C–H and C–C bonds by transition metal centres is of fundamental interest and plays an important role in the synthesis of complex organic molecules from petroleum feedstocks. However, while there are many examples for the oxidative addition of C–H bonds to a metal centre, transformations that feature oxidative addition of C–C bonds are rare. The paucity of transformations that involve the cleavage of C–C rather than C–H bonds is usually attributed to kinetic factors arising from the greater steric hindrance and the directional nature of the spn hybrids that form the C–C bond, and to thermodynamic factors arising from the fact that M–C bonds are weaker than M–H bonds. Not surprisingly, therefore, most examples of C–C bond cleavage either avoid the kinetic limitations by using metal compounds in which the C–C bond is held in close proximity to the metal centre, or avoid the thermodynamic limitations by using organic substrates in which the cleavage is accompanied by either a relief of strain energy or the formation of an aromatic system. It is, therefore, significant that we have demonstrated that W(PMe3)4(h2-CH2PMe2)H is capable of cleaving the C–C bond of quinoxaline (QoxH).
Specifically, rather than tungsten simply coordinating
QoxH in the h6 manner observed for Mo(PMe3)6, the reaction between W(PMe3)4(h2-CH2PMe2)H
and QoxH results in (i) C–C bond
cleavage and dehydrogenation of QoxH to give [k2-C2-C6H4(NC)2]W(PMe3)4,
and (ii) formation of the
dihydride complex (h4-C2N2-QoxH)W(PMe3)3H2.
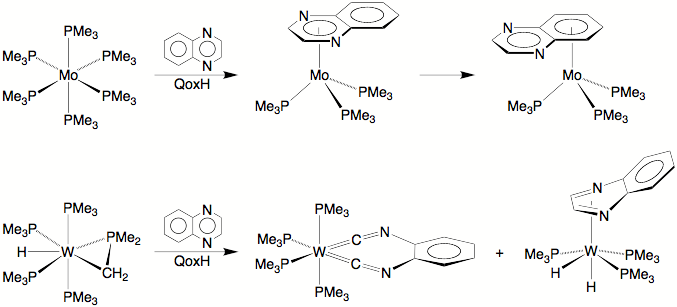
The transformations observed for QoxH are also observed
for 6-methylquinoxaline (QoxMeH) and 6,7-dimethylquinoxaline (QoxMe2H), thereby demonstrating the
generality of the reactions.
The most significant aspect of the reactions between W(PMe3)4(h2-CH2PMe2)H
and quinoxalines pertains to the formation of [k2-C2-C6H2R2(NC)2]W(PMe3)4,
which are the result of breaking an aromatic C–C bond, a bond that is substantially
stronger than a typical C–C single bond. It is also interesting to note that the unprecedented
cleavage of the C–C bond takes place in preference to that of the
C–N bond, a bond that would have been expected to be potentially more
reactive.
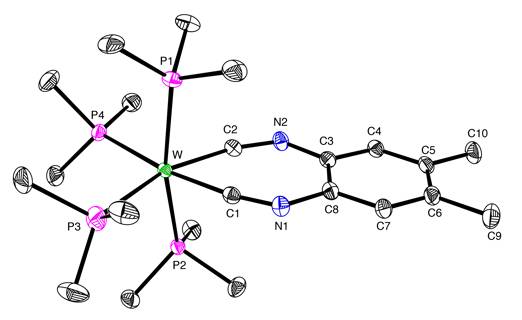
Another interesting feature of [k2-C2-C6H2R2(NC)2]W(PMe3)4 is that chelating isocyanide ligands are not common due to the preference for
MCNR moieties to adopt linear coordination at both nitrogen and carbon. Thus, while o-(CN)2C6H4 has been employed as a ligand, it does not chelate to a single metal, but rather serves as a bridging ligand. Indeed, the smallest ring size
previously reported for a complex of a bidentate di(isocyanide) ligand is 12,
as illustrated by [k2-C2-CH2{OC6H4(NC)}2]Cr(CO)4. Therefore, the 7-membered ring of [k2-C2-C6H4(NC)2]W(PMe3)4 is exceptionally small for a transition metal complex of a bidentate
di(isocyanide) ligand. The ability
to isolate a chelating di(isocyanide) compound with a 7-membered ring may be
attributed to the fact that its mechanism of formation involves insertion of
the tungsten centre into the aromatic nucleus.
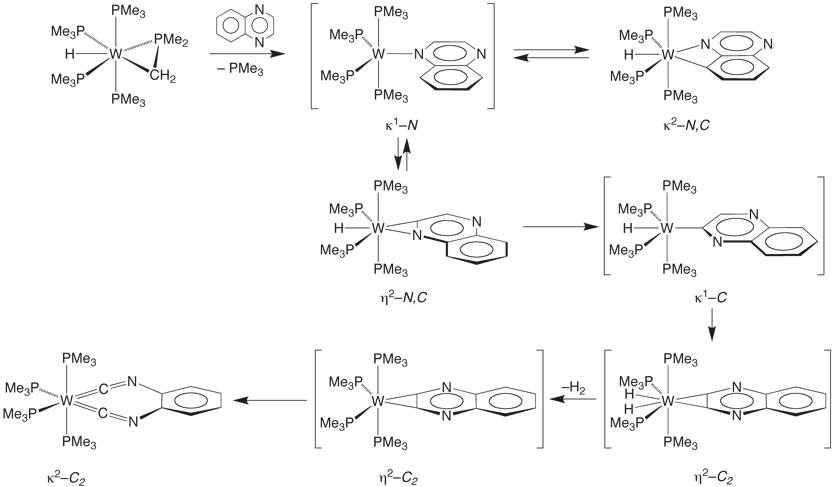
In this regard, the mechanism proposed for cleaving the
C–C bond comprises a series of C–H bond cleavage reactions to
access a benzyne-type intermediate [h2-C2-C6H4(NCCN)]W(PMe3)4H2,
from which [k2-C2-C6H4(NC)2]W(PMe3)4 is obtained via a sequence that
involves reductive elimination of H2 and C–C bond
cleavage. Supporting this
mechanism, evidence that C–H bond cleavage is facile in this system is
provided by the fact that several other compounds, including mononuclear (h2-N,C-Qox)W(PMe3)4H and (k2-N,C-Qox)W(PMe3)4H, are observed
during the course of the reaction.
Selected References
“Cleaving Carbon–Carbon Bonds by Inserting Tungsten into Unstrained Aromatic Rings.” Aaron Sattler and Gerard Parkin Nature 2010, 463, 523-526.
|
|||