Hydrodesulfurization and
Hydrodenitrogenation
Hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) are the processes by which sulfur and nitrogen–containing impurities are removed from crude petroleum feedstocks and fuels, and thereby comprise the largest volume and most important industrial catalytic application of transition metals.
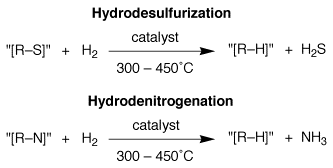
The importance of these processes is twofold: firstly, the sulfur and nitrogen–containing impurities in hydrocarbon fuels have a severe environmental impact resulting from the contribution of sulfur and nitrogen oxides (produced during combustion) to acid rain; and secondly, the sulfur and nitrogen–containing impurities are effective catalyst poisons that prevent crude feedstocks from being used for subsequent chemical transformations.
HDS and HDN catalysts are heterogeneous materials that are commonly referred to as “promoted Mo or W catalysts”. For example, the most commonly employed hydrodesulfurization catalyst is a “cobalt promoted” molybdenum catalyst (“Co–Mo”) comprised of a mixture of MoS2 and Co9S8 supported on Al2O3. However, despite the fact that molybdenum is the most essential transition metal component of a typical HDS catalyst, the majority of homogeneous model studies has focused on metals other than molybdenum. For this reason, we have initiated a series of studies directed towards defining the coordination chemistry of molybdenum relevant to HDS and HDN. These investigations have resulted in the first example of C–S bond cleavage of thiophene by molybdenum and the first demonstration of catalytic hydrogenation of quinoline (and related N–heterocyclic nitrogen compounds) by molybdenum complexes.
C–S Bond Cleavage of Thiophene by Ansa Molybdenocene Compounds
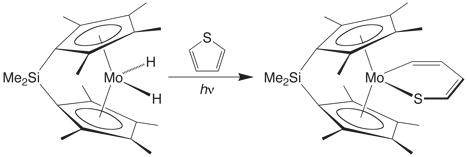
The ansa molybdenocene dihydride complex [Me2Si(C5Me4)2]MoH2 photochemically achieves cleavage of the C–S bond of thiophene to give [Me2Si(C5Me4)2]Mo(h2‑C,S‑SC4H4). Such reactivity is noteworthy because the related non–ansa molybdenocene compounds Cp2MoH2 and Cp*2MoH2 do not achieve C–S bond cleavage under these conditions.
h5–Coordination
and C–S Bond Cleavage of Thiophene by Mo(PMe3)6
Thiophene reacts with Mo(PMe3)6 at room temperature to yield the thiophene adduct (h5–C4H4S)Mo(PMe3)3 and the butadiene–thiolate complex (h5–C4H5S)Mo(PMe3)2(h2–CH2PMe2), with (h5–C4H4S)Mo(PMe3)3 being the first mononuclear h5–thiophene molybdenum complex to be spectroscopically identified.
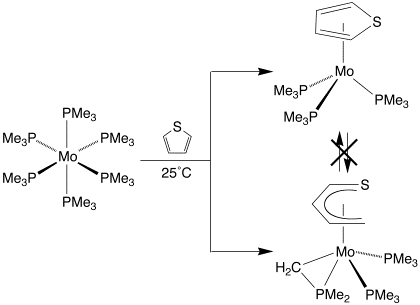
The formation of (h5–C4H5S)Mo(PMe3)2(h2–CH2PMe2) is a result of C–S bond cleavage and hydrogenation of the thiophene ligand. Interestingly, (h5–C4H4S)Mo(PMe3)3 and (h5–C4H5S)Mo(PMe3)2(h2–CH2PMe2) do not interconvert on the time-scale of the experiment, thereby providing evidence that h5–thiophene coordination does not necessarily facilitate C–S cleavage and that such adducts may merely represent resting states during HDS.
Reactivity of Mo(PMe3)6 towards
Selenophene
The reactivity of Mo(PMe3)6 towards selenophene derivatives is pertinent towards understanding the mechanisms of HDS because the products obtained could correspond to different stages of the reaction coordinate for the thiophene system. As such, an analysis of the reactivity of the selenophene systems furnishes insight into the types of transformations that may be achieved by molybdenum as a component of a HDS catalyst surface. It is, therefore, interesting that Mo(PMe3)6 reacts with selenophene to give the metallacyclopentadiene complex [(k2–C4H4)Mo(PMe3)3(Se)]2[Mo(PMe3)4] in which the selenium has been completely abstracted from the selenophene moiety.
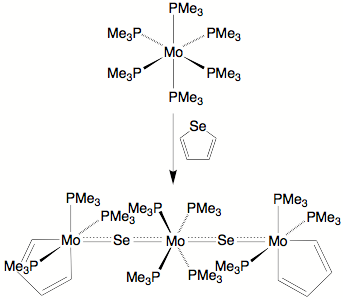
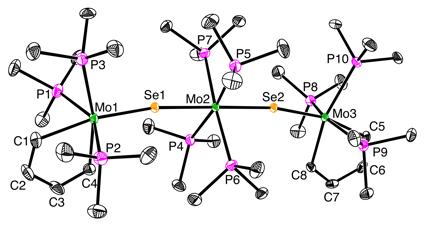
Molecular
structure of [(k2–C4H4)Mo(PMe3)3(Se)]2[Mo(PMe3)4]
Reactivity of Mo(PMe3)6 towards Benzothiophene and Benzoselenophene
The reaction of Mo(PMe3)6 with benzothiophene follows a different course to that of thiophene, thereby increasing our knowledge of the array of organic species that may exist and interconvert on the surface of an HDS catalyst. Specifically, Mo(PMe3)6 cleaves the C–S bond of benzothiophene to give paramagnetic (k2–CHCHC6H4S)Mo(PMe3)4, which rapidly isomerizes to the olefin–thiophenolate and 1–metallacyclopropene-thiophenolate complexes, (k1,h2–CH2CHC6H4S)Mo(PMe3)3(h2–CH2PMe2) and (k1,h2–CH2CC6H4S)Mo(PMe3)4.
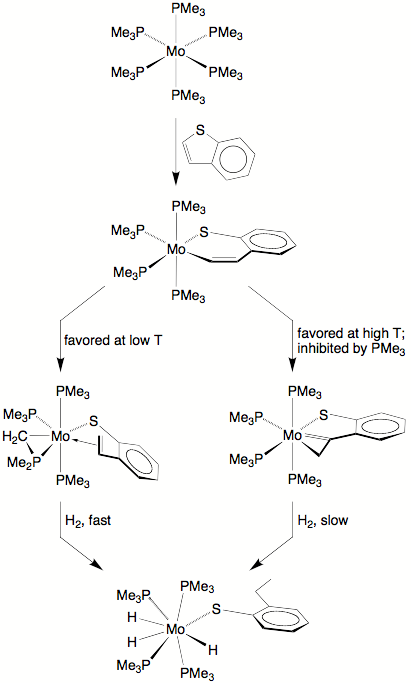
Both (k1,h2–CH2CHC6H4S)Mo(PMe3)3(h2–CH2PMe2) and (k1,h2–CH2CC6H4S)Mo(PMe3)4 react with H2 at room temperature to yield Mo(PMe3)4(SC6H4Et)H3, thereby achieving partial hydrogenation of benzothiophene on a molybdenum center. The relative reactivity of the two isomers, however, differ considerably. Thus, whereas hydrogenation of the former occurs within minutes, hydrogenation of the latter requires a period of days to proceed to completion.
The reaction of Mo(PMe3)6 with benzoselenophene is considerably more complex than the reaction with benzothiophene, with four products having been isolated. Thus, in addition to (k1,h2–CH2CHC6H4Se)Mo(PMe3)3(h2–CH2PMe2) and (k1,h2–CH2CC6H4Se)Mo(PMe3)4, which correspond to two of the species observed in the benzothiophene reaction, products resulting from C–C coupling, namely [k2,h4–Se(C6H4)(CH)4(C6H4)Se]Mo(PMe3)2 and [m–Se(C6H4)(CH)C(CH)2(C6H4)](m–Se)[Mo(PMe3)2][Mo(PMe3)2H], are also formed.
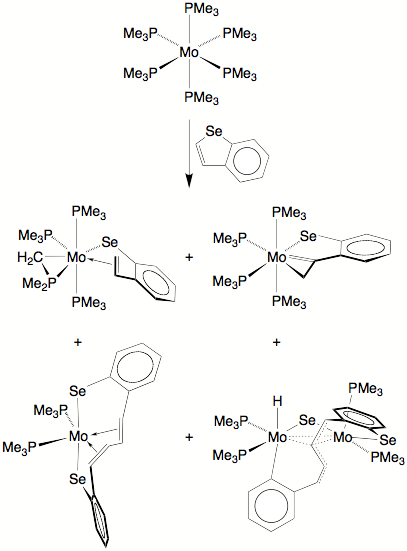
Coordination Chemistry of N–Heterocyclic Aromatic Hydrocarbons with
Molybdenum
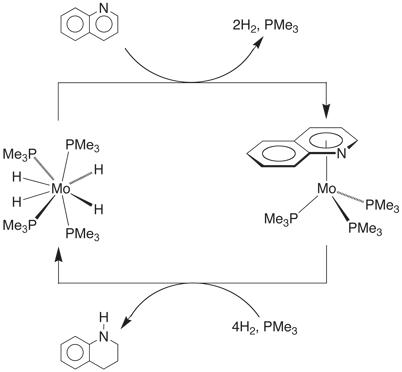
The reactions of the 6-membered heterocyclic nitrogen compounds, pyridine, quinoline, and acridine towards Mo(PMe3)6 are interesting because each follows a different course. Pyridine reacts with Mo(PMe3)6 to give an h2–pyridyl derivative [h2–(C5H4N)]Mo(PMe3)4H as a result of a–C–H bond cleavage. In contrast, the corresponding reactions of quinoline and acridine give products of the type (h6–ArH)Mo(PMe3)3 in which both ligands coordinate in an h6–manner, rather than the more commonly observed h1–coordination mode via the nitrogen atom.
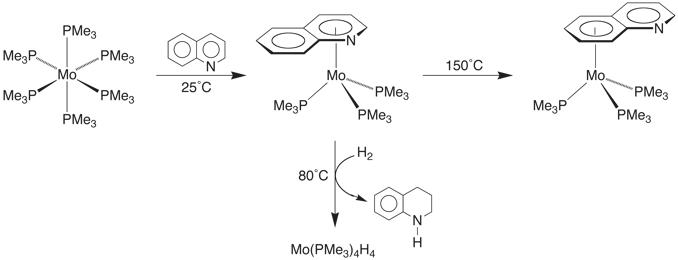
The isolation of the [h6–(C5N)–quinoline]Mo(PMe3)3 isomer is particularly significant because coordination through the heterocyclic ring of quinoline is unprecedented. In this regard, [h6–(C5N)–quinoline]Mo(PMe3)3 is converted to the thermodynamically more stable carbocyclic ring coordinated isomer [h6–(C6)–quinoline]Mo(PMe3)3 at 150˚C. Interestingly, the reactivity of [h6–(C5N)–quinoline]Mo(PMe3)3 and [h6–(C6)–quinoline]Mo(PMe3)3 are quite distinct. Specifically, [h6–(C5N)–quinoline]Mo(PMe3)3 reacts with H2 at 80˚C to liberate tetrahydroquinoline, whereas [h6–(C6)–quinoline]Mo(PMe3)3 is inert under these conditions.
Mo(PMe3)4H4 is the
principal molybdenum-containing product of the hydrogenation reaction, and
since Mo(PMe3)4H4 also reacts with quinoline
to give [h6–(C5N)–quinoline]Mo(PMe3)3, it
is evident that Mo(PMe3)4H4 could, in
principle, serve as a hydrogenation catalyst. In fact, Mo(PMe3)4H4 is the
first molybdenum compound that serves as a homogeneous hydrogenation catalyst
for quinoline.
Recent studies have demonstrated that polynuclear
aromatic compounds inhibit both hydrodenitrogenation and
hydrodesulfurization. For this
reason, it is particularly pertinent to investigate the means by which these
compounds interact with molybdenum. In this regard, while anthracene (AnH) and acridine (AcrH) react with
Mo(PMe3)6 to give (h6-AnH)Mo(PMe3)3 and (h6-C6-AcrH)Mo(PMe3)3,
respectively, in which the aromatic ligands coordinate in an h6 manner via one of the outer rings, the
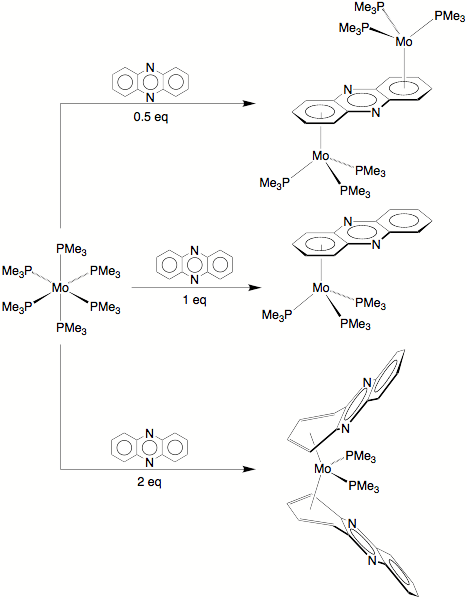
corresponding reaction of phenazine, a related trinuclear heterocycle, is
considerably more complex and results in compounds that exhibit a variety of
coordination modes.
Specifically, in addition to forming (h6-C6-PhzH)Mo(PMe3)3, Mo(PMe3)6 reacts with phenazine to yield (i) (h4-C4-PhzH)2Mo(PMe3)2,
in which two phenazine ligands coordinate via h4–coordination
modes, and (ii) (u-h6,h6-PhzH)[Mo(PMe3)3]2,
a dinuclear compound in which the phenazine ligand bridges two metal
centers.
Selected References
“Modeling Aspects of Hydrodesulfurization at Molybdenum: Carbon–Sulfur Bond Cleavage of Thiophenes by Ansa Molybdenocene Complexes.” David G. Churchill, Brian M. Bridgewater, and Gerard Parkin J. Am. Chem. Soc. 2000, 122, 178-179.
“The Reactivity of Mo(PMe3)6 towards Heterocyclic Nitrogen Compounds: Transformations Relevant to Hydrodenitrogenation.” Guang Zhu, Joseph M. Tanski, David G. Churchill, Kevin E. Janak and Gerard Parkin J. Am. Chem. Soc. 2002, 124, 13658-13659.
“New Modes for Coordination of Aromatic Heterocyclic
Nitrogen Compounds to Molybdenum: Catalytic Hydrogenation of Quinoline, Isoquinoline, and
Quinoxaline by Mo(PMe3)4H4.” Guang Zhu, Keliang Pang and Gerard
Parkin J. Am. Chem. Soc. 2008, 130, 1564-1565.
“Reactivity of
Mo(PMe3)6 Towards Benzothiophene and Selenophenes: New Pathways Relevant to
Hydrodesulfurization.” Daniela
Buccella, Kevin E. Janak and Gerard Parkin J. Am. Chem. Soc. 2008, 130, 16187–16189.
“Multiple Modes for Coordination of Phenazine to Molybdenum: Ring Fusion Promotes Access to h4-Coordination, Oxidative Addition of Dihydrogen and Hydrogenation of Aromatic Nitrogen Compounds.” Aaron Sattler, Guang Zhu, and Gerard Parkin J. Am. Chem. Soc. J. Am. Chem. Soc. 2009, 131, 7828-7838.
|
|||